
Comparing classic plasma fractionation separation methods—and what comes next.
Read More
Manufacturers are poised to take advantage of the growth of the antibody-drug conjugate (ADC) market. To do so they need to understand the process technologies involved, as well as the manufacturing challenges.
The concept of a “magic bullet”—a way to deliver deadly toxins directly to a pathogen or cancer cells—originated with Paul Erlich in the early 1900s. He imagined a molecule capable of targeting the pathogen or cancer cells, a toxin, and a way to link the two. Almost 100 years later Erlich’s vision came to fruition when the first ADC received FDA approval in 2000.
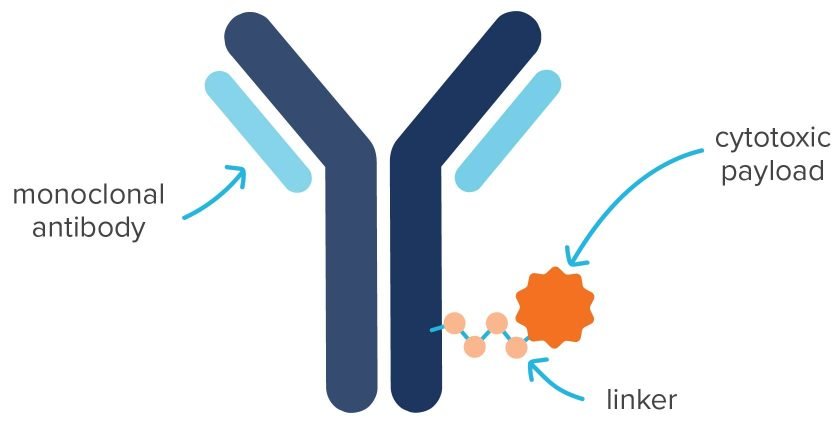
ADCs are targeted biopharmaceuticals, consisting of a monoclonal antibody (mAb) covalently bound via a chemical linker to a cytotoxin, typically a small molecule drug. All of the currently approved ADCs are oncology treatments, with breast cancer therapies dominating the market with the largest revenue share in 2022. They are also in development to treat fungal and bacterial infections and to make synthetic vaccines. This article is focused primarily on highly potent cytotoxic compounds as the drug payload.
These targeted therapies are needed to overcome some of the limitations of standard oncology treatments. These include the inability to surgically access all sites, the systemic effects of traditional chemotherapy, and the peripheral damage caused by radiotherapy. In addition, targeted therapies using mAbs alone are not always able to fully destroy cancer cells.
ADCs combine the power of chemotherapy with the precision of using antibodies to target cancer cells while minimizing systemic toxicity and other off-target effects. The aim is to develop ADCs that increase the therapeutic window (i.e., the range of dosages producing maximum benefit with minimal side effects). Despite this, ADCs can still exhibit a high incidence of adverse events, including neuropathy and toxicity in the blood, liver, eyes, or GI tract. This is due to expression of the target antigen on normal cells, premature release of the payload, or binding of the ADC to similar targets expressed on healthy tissue.
There has been considerable growth in the ADC market in the past five years due to a focus on antineoplastic payloads, improvements to payload linker stability, improved testing for antigen expression, ability to identify clean targets, and the use of data to analyze and develop more effective combinations. We are currently in the third generation of ADC deployment, which began in 2017 with the reapproval of Mylotarg and has been followed by an explosion of approvals and clinical trials.
We are moving into the next generation of ADC development, with ongoing research into multi-arm linkers and conjugation with glucagon-like peptide-1 (GLP-1), as well as non-traditional ADCs, including radionuclide ADCs (RDCs), bispecific conjugates, antibody-fragment drug conjugates, and immune-stimulating antibody conjugates (ISAC). Conjugates are in development that use oligonucleotides instead of antibodies as the targeting portion of the conjugate or as the payload in the form of antibody-oligonucleotide conjugates (AOCs). C’Dot drug conjugates (CDCs) and peptide drug conjugates (PDCs) are interesting technologies able to cross the blood-brain barrier.
There are currently 13 FDA-approved ADCs on the market. More detail, as well as a list of linkers and cytotoxic drugs used in FDA-approved ADCs can be found here in the National Library of Medicine..
Mechanism of action
ADCs work through various mechanisms of action (MOA). In payload-driven cell death, the mAb portion of the ADC recognizes and binds to an antigen uniquely expressed on the surface of cancer cells. Once bound, the ADC is internalized via endocytosis. With ADCs using non-reducible/non-cleavable linkers, the mAb and linker are degraded in the lysosome by proteolysis, releasing the payload. Lysosomal degradation releases the cytotoxin, whereupon it exerts its toxic effects. This is precision medicine delivered to tumor cells while limiting off-target effects.
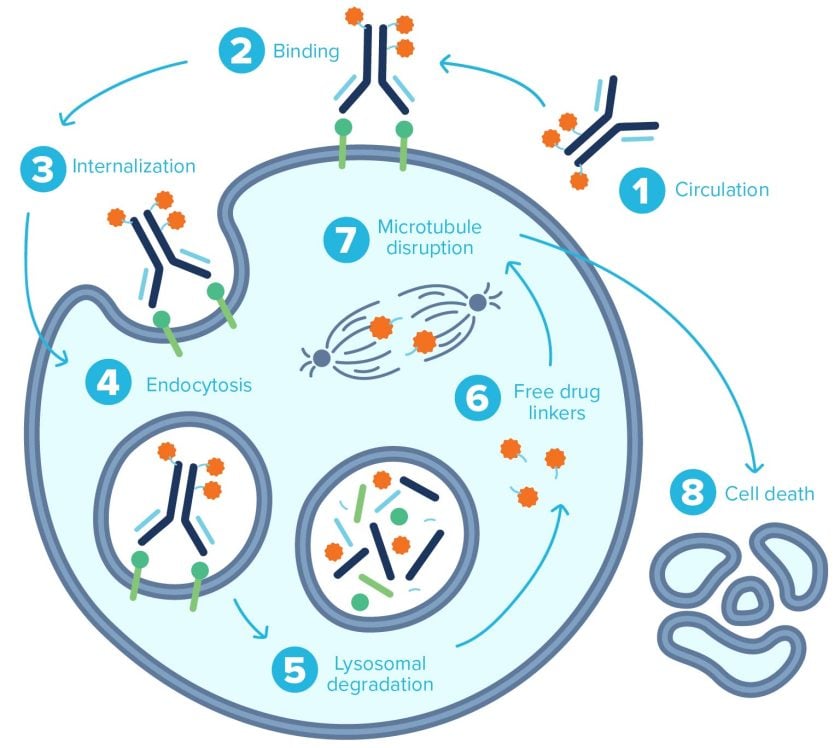
Microtubule inhibitors
Trastuzumab emtansine (Kadcyla), used for the treatment of HER2-positive breast cancer, is an example of an ADC that includes a microtubule inhibitor as its payload. The mAb (trastuzumab, or Herceptin) binds to the HER2 receptors present on the surface of these breast cancer cells, triggering endocytosis. The ADC is degraded within lysosomes, releasing free DM1, a cytotoxin that inhibits microtubule assembly and leads to cell death.
DNA alkylating agents
Loncastuximab tesirine (Zynlonta) is used to treat large B-cell lymphoma. The mAb is linked to the cytotoxic drug, SG3199, which alkylates and cross-links DNA and prevents cell division.
RNA polymerase II inhibitors
Inhibition of transcription prevents protein synthesis and leads to cell death. There are promising preliminary studies suggesting RNA polymerase II inhibitors can be used as cytotoxins. However, there are no FDA-approved ADCs on the market yet.
Topoisomerase I inhibitors
Trastuzumab deruxtecan (Enhertu) is used to treat various cancers. It contains the cytotoxin, deruxtecan, which inhibits topoisomerase I activity via intercalation of DNA.
Definitions
Each mAb is a protein that binds to a specific antigen in the body and is produced by a clonal cell line. This acts as the delivery vehicle for ADCs.
A toxic substance that is attached to the mAb. Also known as the drug, payload, warhead, or highly potent API (HPAPI). The most prevalent targets of a cytotoxin are DNA and tubulin. An example is monomethyl auristatin E (MMAE).
A molecule connecting the cytotoxin to the mAb. Effective linkers exhibit high plasma stability to avoid premature release of the cytotoxin. Cleavable linkers rely on the physiological environment for release of the cytotoxin. Non-cleavable linkers have a non-reducible bond to an amino acid residue in the mAb, which makes them more stable in blood. An example is a thioether, which is dependent on lysosomal degradation of mAb to release the cytotoxin. An example is SMCC, which links a highly potent drug (emtansine or DM-1) to a mAb (Herceptin) to produce the anticancer drug, trastuzumab emtansine (Kadcyla). Some newer ADCs (e.g., Lumoxiti) have no linker—an antibody fragment is covalently bound directly to a cytotoxin.
Any substance that stimulates an immune response. External antigens include components of, or substances produced by, viruses (e.g., the SARS-CoV-2 spike protein) and microbes (e.g., bacteria, fungi, protozoa), toxins (e.g., snake venom), or any other foreign agent in the blood.
Autoantigens, which originate within the body in those with autoimmune disorders, provoke an immune response that creates autoantibodies.
The ratio of the number of molecules of cytotoxins per antibody. The usual therapeutic range for the DAR is 2–8.
Process technologies used in ADC manufacturing
Like the molecules themselves, the processing steps to make an ADC are more complex than for most other drugs. Conjugation of the mAb to the cytotoxin is complicated by the low occupational exposure limit (OEL) of the raw materials and final product. The removal of impurities—non-conjugated, semi-conjugated, or aggregated materials among them—can be difficult, given the need for a drug product with the correct DAR and lacking any free payload.
Advances in conjugation technology have allowed for improvements in the ability to modulate DAR and conjugate the payload to specific sites on the mAb. Conjugation site specificity has been shown to impact thermal stability, aggregation, ADC metabolism, and stability in circulation. It also simplifies analytics and improves therapeutic index. With conjugation improvements and expectations for homogeneity, additional purification technologies are needed.
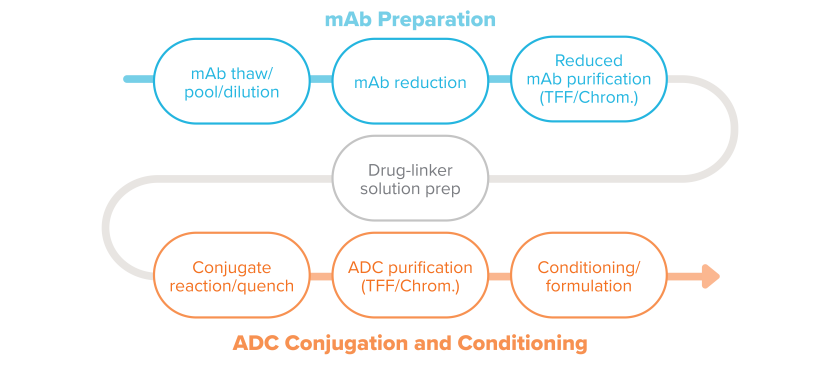
An example of a typical conjugation process in ADC manufacturing.
Here are descriptions for the typical conjugation process steps.
1. Thaw bulk mAb
The process for making ADCs begins with the raw materials, the manufactured mAb, and payload-linker. The frozen bulk mAb is thawed and pooled as needed.
2. Buffer exchange and mAb dilution
Buffer exchange, when needed, uses tangential flow filtration (TFF). Bulk mAb is then added to the modification reactor to prepare it to receive the linker. The correct concentration of mAb is obtained through dilution with various buffers.
3. mAb modification
There are a number of methods for attaching the linker to the mAb. The mAb preparation may include reduction and reoxidation steps. Modification of the mAb is particularly important when working with site-specific conjugation processes to open and prepare the conjugation sites. Contact time with reagents, reagent concentration, and temperature are critical process parameters during these steps.
4. Modified mAb purification
Purification of the modified mAb typically includes TFF or chromatography as seen in standard mAb manufacturing processes. The purification processes are implemented to remove reaction reagents, remove high molecular weight molecules (aggregates) if present, exchange buffers, as well as achieve the appropriate mAb concentration for the conjugation reaction.
5. Drug-linker dissolution
The cytotoxic-linker combination, a highly potent active pharmaceutical ingredient (HPAPI), is weighed and dispensed in an isolator and put in solution. The cytotoxic-linker combination is typically a hydrophobic compound and requires a non-aqueous co-solvent for full dissolution. In addition to the primary containment equipment, the low OEL of these HPAPIs—which can be as low as picograms per cubic meter— often dictates use of powered air purifying respirators (PAPR) or breathing air at all times when handling cytotoxins. Gowning and other personal protective equipment may also be used to provide protection in potential upset conditions. Cytotoxins are at their greatest hazard when they are in powder form during weighing and, later, lyophilization of the ADC. It’s assumed the drug and linker are received as one molecule, referred to here as the cytotoxin.
6. Conjugation reaction
Drug-linker solution is transferred to the conjugation reactor at a specified rate to control temperature increase due to heat of dilution. The bioconjugation of the drug-linker to the mAb tends to be a small-scale reaction, producing gram to low-kilogram quantities of ADCs for clinical trials. Some commercial processes use reactors as large as 1,200 L, with ongoing studies to develop 2,000 L reactors.
Temperature, pH, reaction duration, and drug-linker solvent concentration are critical parameters to ensure efficient reactions and consistent quality. Due to the crossover of scientific disciplines with respect to analytical methods, safe handling practices, and conjugation technologies, biologists and chemists must be partners in both product and process development.
7. Purification with ultrafiltration/diafiltration (UF/DF) or chromatography
Purification of the complete ADC uses TFF, chromatography, or a combination to remove free payload, linkers, and mAbs, ADCs with incomplete DAR, agglomerates, and other impurities. It also removes the solvents, including such compounds as dimethyl sulfoxide (DMSO), ethanol, and dimethylacetamide (DMA). Other purification methods are in development, including size-exclusion chromatography, multi-column chromatography, continuous chromatography, and hydrophobic interaction chromatography (HIC).
8. Formulation of bulk drug substance (BDS)
In some cases formulation involves diluting the drug substance in buffer, then freezing. The BDS then goes on to the fill-finish step in the process.
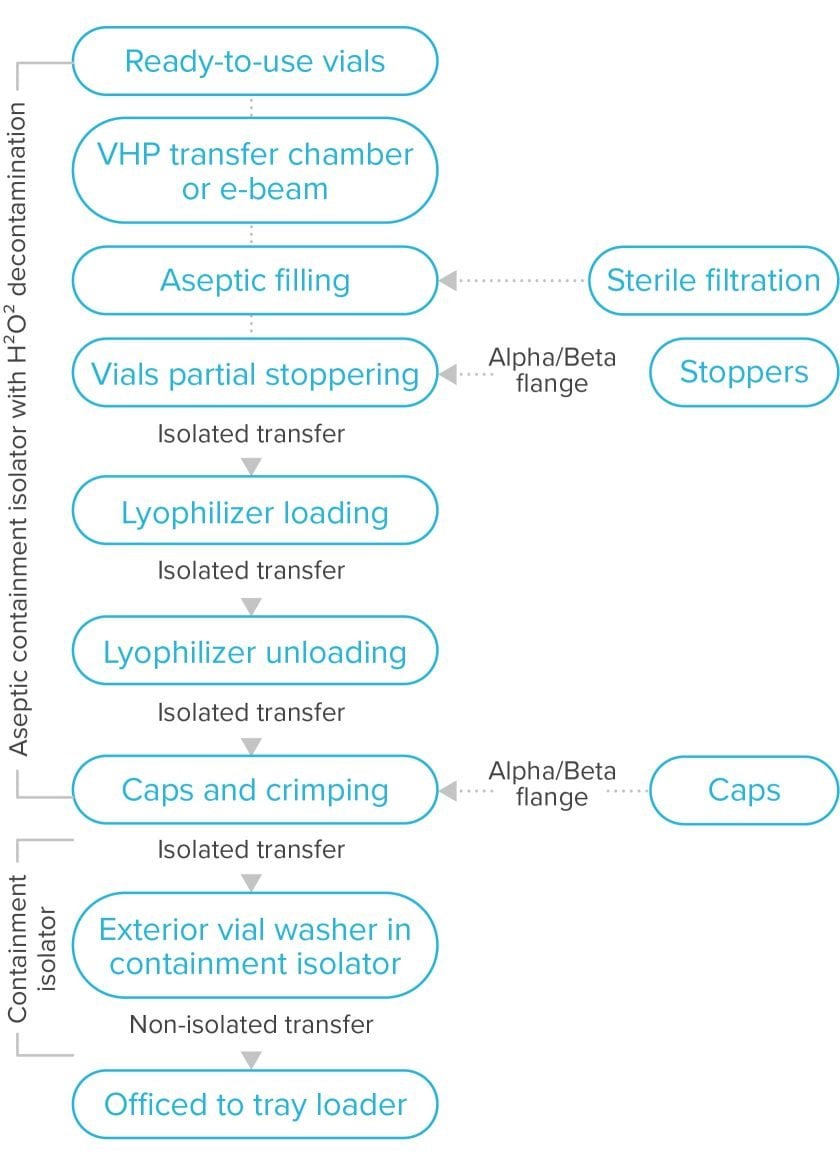
The fill-finish step
This is similar to a standard fill-finish process with some exceptions due to the toxicity of products. The filling system includes isolated, semi-automatic, or automatic vial filling auto-loaded into a lyophilizer to prevent worker exposure. Since the most dangerous form of a cytotoxin is the dry form, the most critical steps to prevent exposure are weighing and dispensing (at the beginning of the process) and handling of the lyophilized ADC (at the end). Once the vial has been capped and crimped, it’s transferred to an external vial washer, housed in another containment isolator, to protect anyone handling the vial downstream. Exterior vial washers are unique to this type of fill line and are not normally seen in a standard biotech fill line.
Lyophilization
Due to the stability of the conjugated product, most commercial formulations (>90%) are presented in lyophilized vial format. While people are working on improving formulation stability, there’s no push to remove lyophilization in the near future. For early stage products, such as those in clinical development and trials, products may be delivered in liquid format.
In addition to Annex 1 requirements ensuring lyophilizer sterilization, ADC lyophilizers need automated cleaning capabilities to prevent cross-contamination and employee exposure during equipment inspection. The waste cleaning solutions must be collected and treated appropriately to keep potent material out of the local water system.
ADC Manufacturing
Many of the challenges faced by ADC manufacturers are largely the same ones throughout the industry—to provide consistently pure and efficacious drugs free of environmental and process-related contamination. However, given that development of the current generation of ADCs focuses on the activity and specificity of the drugs, as well as the toxic nature of these highly complex molecules, specialized manufacturing techniques are necessary. Changes have occurred in the past decade, particularly to meet the safety challenges unique to ADCs, both in terms of product and personnel. This includes risk assessment, changes to materials and equipment, improvement in facility design, and changes to cleaning practices.
To be competitive, manufacturers need to operate facilities dedicated to high-potency biologicals that:
- Keep pace with technological advances
- Have well-established platform operations
- Have a skilled workforce with the specialized expertise to meet the expectations of customers and regulators
- Possess novel analytical (technologies/processes) capable of in-depth molecular analysis
Lack of adequate ADC manufacturing capacity
The rapid expansion of approvals and demand for ADC treatments has put pressure on manufacturing capacity already tapped out making essential products for the COVID-19 pandemic, as well as to fuel the burgeoning markets for other biologics. The complexity of ADCs depends on uniquely designed and equipped facilities—a company with a high-speed, high-throughput process for making a vaccine, for example, can’t simply pivot to manufacture ADCs. The production of these innovative products is more complex at every stage, including formulation development, the supply chain, manufacturing, and fill-finish.
Given this, CMOs and CDMOs have rushed in to meet the need, and many manufacturers are turning to CMOs to increase speed to market. This was evidenced by the 59% of life sciences experts we surveyed in our report, Horizons: 2022 Life Sciences, who said they intended to use CMO services in the next five years to expand production.
Equipment
Some equipment used to produce ADCs is different, with additional requirements, and this can increase costs. Product instability can be problematic. In solution, a linker can become unbound, giving rise to free payload. Lyophilization greatly improves product stability. Advanced purification methods are also needed to remove free payload in solution and other impurities.
Challenges with TFF
TFF is used to overcome product instability, to remove free payload and free linker, and concentrate the ADC for downstream purification steps. Attention to the concentration on the upstream side of the membrane, as well as ensuring adequate permeate flow, are two critical pieces to the ADC purification puzzle. The ideal membrane size needs to be determined in process development.
Standard TFF membranes can also pose a risk when operators are changing TFF membranes. As a contaminated piece of equipment, if they can clean and decontaminate it post use, then they can change the membranes in an open room. If they cannot decontaminate it, then we have to look at methods to contain the membrane and its holder while changing out the membrane. There are capsules on the market that can contain the membrane and holder during change outs, preventing exposed ports and material.
Weigh-dispense isolator design questions
You need to look at how the primary containment is used and whether you need to access the internals of that isolator multiple times. Often, there’s a rapid transfer port (RTP) to ensure safe movement of material from the isolator to other equipment or a waste shoot. Do you need a single chamber that you’ll only access once or a dual-chamber system so you can open and close a port multiple times and attach multiple components? This needs to be resolved in the design phase.
Significant cost increases due to isolator technology
Risk mitigation during filling can lead to significant cost increases compared to a standard filling line without HPAPIs. Not only do the number and type of isolators change, but this leads to other design and equipment changes, including increased HVAC requirements and the need to eliminate bubbles and sinks at in-feed and out-feed. A $12 million, 200 vials per minute line can balloon to $24 million for an ADC.
Cleaning and cross-contamination control
The toxic nature of these products demands a balance of containment, facility and equipment cleaning, and aseptic design for the process and the entire facility. This is critical to safety and limits the exposure of personnel, not just in the room, but in the entire facility, which may be a multi-product facility that uses the same equipment for different products. There’s a lag in the creation of regulatory guidance for the production of ADCs. Instead, manufacturers are expected to follow FDA guidelines that cover mAbs, as well as those for small molecules under combination product requirements.
Workplace health and safety
In addition to guidelines from the FDA and other regulators, the toxicity of ADCs and the raw materials means following safety guidelines from the Occupational Health and Safety Administration (OSHA), the National Institute for Occupational Safety and Health (NIOSH), and exposure requirements from the CDC. Training includes identification of toxins, the risks of exposure, and cleanup procedures. The vast majority of industry experts surveyed (92%) said their sites use an OEL threshold for ADC production of at least 100 ng.
Success depends on working closely with both regulators and subject matter experts to keep pace with rapid innovations. Equipment technology is advancing and there are opportunities to adopt innovations being applied in other areas, such as the protection needed throughout the radiopharmaceutical supply chain. Bringing regulators in at the start of a project helps them understand why you’re making certain decisions.
It’s possible to use fixed and dedicated equipment, but you’ll need to ensure complete removal of all contamination after each run. Some use glass and stainless steel conjugation reactors, then discard them because they can’t confirm removal of all product from the equipment. However, advancements in detection methods are allowing companies to develop multi-product facilities with thorough cleaning and sampling regimes. Single-use equipment has advantages with respect to reduced cross-contamination risks but comes with compatibility challenges due to interactions between solvents used in dissolution and conjugation steps and the USP Class VI elastomers used in the equipment. Recent improvements in film material strength, closed-capsule TFF membranes, and single-use instrumentation have elevated the viability of single-use systems in the more hazardous areas of the ADC manufacturing process.
Primary containment
OSHA requirements and CDC recommendations for exposure control stress that removal of the source of contamination is the first line of defense. Since this isn’t feasible for ADC production, primary containment (point source containment) is used to protect operators and those outside the production space. It includes the use of isolators, split butterfly valves with exhaust extraction or cleaning the outside of exposed surfaces of the valves prior to disconnection, and, in general, closed processes.
Secondary containment
Secondary containment is added to protect against accidental release of cytotoxins. For process equipment, this includes use of a closed vessel as the primary containment for a process, then putting that in an isolator (secondary containment), biosafety cabinet, or hood. Most still rely on direct protection of operators, by using PAPR and gowning. Protection of the facility relies on design elements, including managing airflow, air locks on entrances, and HEPA filtration on room returns and feeds.
Complex supply chain
Sourcing raw materials, including mAbs and HPAPIs, depends on working with multiple suppliers, multiple quality systems, and tight management of lead times. International transfer of cytotoxins is complicated by complex regulations and paperwork. For these reasons, it can be beneficial to streamline management of the supply chain and, perhaps, bring the supply chain in house.
Scale up
The transition from clinical to commercial ADC manufacturing is significant because, not only does the number of manufactured doses increase, there are changes to equipment, facility design, analytical methods, and cleaning requirements. Commercial manufacturing requires more efficient processes and movement of material and personnel. It depends on an understanding of relevant building codes and OSHA guidelines. All of these depend on working with process development experts who can identify and prepare for the pitfalls from moving small-scale to commercial manufacturing.
Processes used to make small amounts of an ADC in the lab can be scaled out by duplicating equipment, but not easily scaled up. For example, ultracentrifuges, which are often used as part of purification during process development, can be scaled out but not up. Such a process cannot be optimized if it’s run in a suite full of expensive centrifuges with isolators around them. Because of this, the small-scale purification processes developed in a lab need to be scaled up for production runs on different equipment, typically TFF and chromatography equipment. UF/DF is also typically not used in the lab, but required for purification at larger scales.
Analytics
Analytical methods are used to test the drug product for purity and homogeneity, and to ensure the drug container and equipment used to produce the drug are free from contamination. Given that small-scale production for many relies on a CDMO partner, technology transfer and verification methods are needed. Depending on the contract structure, the product sponsor may not have access to much of the analytical methods IP. Good communication, contract structure, and possibly, an in-person transfer are recommended.
Each cytotoxin has detection methods for determining whether it or ADCs are contaminating equipment, room surfaces, or the air. Personnel protection relies on these analytical tools and are used to assess whether equipment has been adequately decontaminated. Additionally, the container/vial itself needs to be contaminant-free to protect handlers downstream, including patients.
Manufacturers need to be prepared for the high cost of some analytical equipment, such as a liquid chromatography mass spectrometer.
Waste handling
ADC manufacturing involves weighing, dispensing, and handling toxic material. Single-use processes generate considerable solid waste, including worker PAPR and single-use isolators, while stainless steel processes create a lot of liquid waste. These processes result in significant amounts of hazardous waste, all of which needs to be handled and disposed of, possibly with onsite incineration. Taking into account the industry-wide focus on reducing waste and environmental sustainability, decisions about how to handle both solid and liquid waste include whether to incinerate, whether this occurs onsite or offsite, whether clean-in-place flushes should be collected, and how to deal with the environmental impact of the materials used (e.g., solvents).
Simplifying your ADC manufacturing journey with CRB
ADC manufacturing facilities are challenging to design given the need for containment, cleanliness, and the safety of product and personnel. It can be faster, safer, and more economical to engage experts who understand the differences in facilities and technologies. As well, streamlining your supply chain by in-house vertical integration can be accomplished by partnering with firms that have a thorough understanding of the manufacturing process, cross-discipline collaboration, safety and facility design requirements.
CRB excels at creating this type of partnership with project owners. Our process, design, and construction knowledge can help solve problems you haven’t been able to solve before. Here’s a few of the places we can to support the development and manufacturing of your next ADC project:
- Risk assessments
- HPAPI handling
- Conjugation
- Fill-finish
- Early discussions and knowledge of containment requirements
- Integration of biologics and small molecule product groups
- Hazardous waste handling
Let’s talk about how to smooth the path for your next ADC project.
