When it was first released in the 1970s, Annex 1: Manufacture of Sterile Medicinal Products filled an important gap in the regulatory landscape by formalizing EU regulators’ expectations for maintaining product sterility through facility design and operation.
Five decades and several revisions later, Annex 1 continues to play a key role in the regulatory landscape. Its latest update, released in 2022, is more than forty pages longer than the previous edition, a testament to the scale of change and innovation underway in the life science industry. Amidst that change, one thing remains constant: the goal of Annex 1 is to ensure the safety of sterile and aseptically manufactured products for patients.
To meet that goal, today’s manufacturers need answers to their most pressing Annex 1 questions. Alongside other industry leaders, our team is helping to develop those answers and advise manufacturers as they move forward in this new and complex regulatory landscape.
Annex 1 Application
1. Does Annex 1 apply to the products I manufacture?
2. Does Annex 1 apply in my region?
Annex 1 Gap Analysis
3. How do I know where my Annex 1 gaps are?
4. What are some common Annex 1 compliance gaps?
Next Steps Toward Annex 1 Compliance
5. I found Annex 1 gaps in my operation. What now?
Annex 1 Application
1. Does Annex 1 apply to the products I manufacture?
For some manufacturers, this answer will be very clear. Others face more ambiguity.
YES
Annex 1 applies if you produce the following:
- Sterile or aseptically produced drug products and investigative medicinal products. This includes animal health products sold in the EU.
- Non-drug products that carry a sterile claim, such as:
- Saline, nasal sprays, ophthalmics and other sterile non-drug products
- Medical devices or combination products
- Ready-to-use vials, syringes, stoppers, pre-sterilized filter paths and other sterile supplies
- Although manufacturers of these items may not face direct regulatory inspections, they should prepare for increased scrutiny from their customers, who are under pressure to minimize the risks of particulate, bioburden and endotoxin contamination through strict contamination control strategies.
MAYBE
Though regulators developed Annex 1 with sterile drug products in mind, many of its core principles offer valuable guidance for manufacturers seeking to eliminate contamination risks for products or operations with a low bioburden requirement. Examples include:
- Drug substances
- Low-bioburden liquids, creams and ointments
- Most heat sterilization or dry heat sterilization operations
- Compendial utility systems (i.e. gasses, water, steam)
- Facilities with cleanrooms required for processing medical products
TO BE DETERMINED
It’s unclear whether oral solid dosage products fall under the Annex 1 purview. Manufacturers should consult regulatory experts on a case-by-case basis to make this determination.
Advanced therapy medicinal products (ATMPs) also fall into a gray area.
Because ATMPs are aseptically handled, Annex 1 may apply. However, the European Commission has a stand-alone guidance for ATMPs (EudraLex Volume 4, Part IV: Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products) which clearly states that no other annexes apply to this drug type.
On the other hand, PIC/S frequently references Annex 1 in their ATMP-specific Annex 2A guidance document. Again, a case-by-case assessment by a team that specializes in the intersection between drug development and regulatory considerations is the best way forward. At minimum, manufacturers should perform a gap analysis against Annex 1 and evaluate patient risks in terms of facility design, operation, and manufacturing processes as part of an overall product contamination control strategy.
What is sterile manufacturing?
Manufacturers have two possible pathways to product sterility: terminal sterilization and aseptic processing.
Terminal sterilization is the ideal approach. This step eliminates living microorganisms from the product, either by killing them via heat or chemicals or by using a filter to physically remove them.
Some drug products cannot withstand terminal sterilization. In those cases, manufacturers may rely on aseptic processing to achieve product sterility. An aseptic process eliminates the possibility of contamination through end-to-end risk-based process controls.
2. Does Annex 1 apply in my region?
Annex 1 is enforceable in all 27 national competent authorities of the European Union. The Pharmaceutical Inspection Co-operation Scheme (PIC/S) has also adopted Annex 1, bringing the updated guidelines to all 54 of its regulatory body members (some of those members have formalized Annex 1 as part of their local laws, while others have not).
The World Health Organization (WHO) has also implemented their own adapted version of the EU’s Annex 1, as have regulatory agencies in Saudi Arabia, Canada and the UK.
There are a few countries where Annex 1 is not yet enforceable, such as the US and China. In these areas, regulators have not formally incorporated Annex 1 requirements into their GMPs. This means that manufacturers who make and sell their products exclusively in their own country may not have to meet Annex 1 requirements. However, these manufacturers may choose to align with Annex 1 practices to maintain their competitiveness in a global marketplace that’s heavily influenced by standards from the EU and the WHO.
The impact of Annex 1 in the US
Here are a few reasons why US-based manufacturers may need to meet Annex 1 expectations, even though the guideline has not been formally adopted by the FDA:
If you manufacture products in the US but export them to regions where Annex 1 applies, you must follow Annex 1 guidelines.
Even if you don’t currently export your products but you might in the future, consider familiarizing yourself with Annex 1 and its implications today. Small, intentional changes to your facility and processes now could streamline your Annex 1 compliance process in the future.
FDA regulators may scrutinize your operation through an “Annex 1” lens.
The FDA contributed to the Annex 1 update, and the agency considers itself “generally aligned” with its content.
What does that mean for US-based manufacturers? Even if they don’t export, they should understand and broadly integrate the principles of Annex 1 in order to meet the rigors of cGMP manufacturing. The “c,” after all, stands for “current” and implies alignment with the global industry’s latest standards and expectations.
However, there are a few areas where the FDA’s expectations deviate from the guidelines laid out in Annex 1. In particular:
- ISO vs. Grade and Airlocks
The FDA applies the ISO clean room classification scheme. Classification for “in operation” occupancy states begins with unclassified space then proceeds to ISO 8, ISO 7, ISO 6, and ISO 5, with specific particulate and bioburden requirements for each classification. Isolators must be in ISO 8 backgrounds while in an “in operation” state, according to FDA requirements. One airlock is required between each ISO step.
By contrast, Annex 1 classifies controlled environments using a grade classification scheme. That scheme starts with unclassified and continues to Grade D, Grade C, Grade B, and Grade A. Like ISO classifications, each grade has specific particulate and bioburden requirements for “in operation” occupancy states; however, the Annex 1 grade classification scheme also establishes requirements for cleanrooms that are “at rest.” Open isolators must be in Grade C at a minimum. An airlock is required between each grade.
In practice, this means that a facility designed to meet FDA requirements may only require one set of airlocks to access the isolated filling line room (located between the unclassified and ISO 8 space), while an Annex 1 facility would require two sets of airlocks (one located between the unclassified and Grade D spaces and a second one between the Grade D and Grade C spaces). To comply with both regulatory agencies, a facility would require two airlocks.

- The Contamination Control Strategy (CCS)
Although not strictly required according to US regulations, having a CCS in place will indicate to FDA regulators that you have developed appropriate protocols for de-risking your sterile or aseptic process and ensuring patient safety. In other words, the FDA does not require manufacturers to have a document called a Contamination Control Strategy, but they will expect you to have implemented the risk mitigation strategies and quality systems typically included in a CCS.
- Pre-Use Post-Sterilization Integrity Testing (PUPSIT)
To address concerns that the sterilization process for filters may cause undetected damage, Annex 1 requires PUPSIT. Although some European authorities strictly enforce the use of PUPSIT for all products, others allow manufacturers to prove, through appropriate scientific justification and other risk mitigations, that PUPSIT is not required for their process.
Similarly, manufacturers solely under the FDA’s purview can avoid PUPSIT if they’re able to show, through robust risk-based assessments, that it is not necessary (or that it may introduce more significant risks to patients).
Annex 1 Gap Analysis
3. How do I know where my Annex 1 gaps are?
Manufacturers must undertake a gap assessment process to identify potential Annex 1 compliance issues.
A deep understanding of current regulatory requirements is fundamental to this process. An architecture, engineering and construction (AEC) partner with experience in this area can help, working with manufacturers to identify where current operations fall short of those requirements and evaluate the criticality of any identified gaps. From there, the AEC team can help to prioritize remediation projects and determine associated costs and timelines.
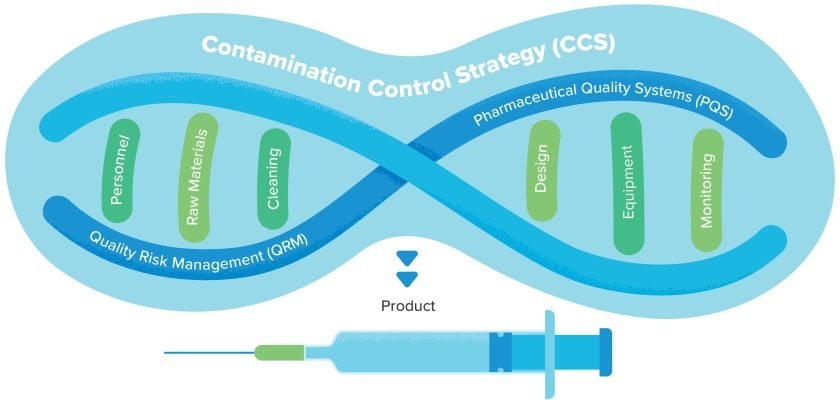
The Contamination Control Strategy: A framework for Annex 1 gap assessments
Previous iterations of the Annex 1 guidance document required manufacturers to control contamination risks, but the latest revision goes further by explicitly requiring a robust CCS. In just 59 pages, the Annex 1 document mentions the CCS more than 50 times—a sign of its importance.
Historically, manufacturers aimed to keep drug products free from microbial, particulate and endotoxin contamination through a combination of SOPs, standalone risk assessment reports and other documents. Many of these documents were typically located in different places and not necessarily standardized across the whole facility, let alone across a global organization. By formalizing the CCS, Annex 1 requires manufacturers to consolidate these documents into a single, continuously evolving centralized framework.
A robust CCS is comprehensive and holistic. By leveraging Quality Risk Management (QRM) and the Pharmaceutical Quality System (PQS), the CCS identifies potential sources of contamination, defines all critical control points, and establishes a process for developing Corrective and Preventive Actions (CAPAs) when necessary. This makes it a vital input during the design process of new facilities and a useful tool when assessing existing operations against Annex 1 requirements.
The CCS Playbook: Scoring early and defending the win!
For all the aseptic manufacturers here, consider this: if your operation were a hockey game, you would be under pressure to score all your goals in the first period by taking the necessary steps to establish sterility. After that, you’re playing defense for the rest of the game, protecting your “win” by closely monitoring the manufacturing process and controlling all contamination risks.
In this metaphor, the CCS is your game plan, providing the overall strategy you need to score goals early and maintain your lead until the final whistle blows.
During the gap assessment process
Large manufacturers often have an in-house team tasked with identifying and documenting Annex 1 gaps. Many turn to third-party experts for assistance.
Some of those independent teams approach the gap assessment process like a regulating agency: they passively observe your operation, then submit a report based on what they see. Others take a more embedded approach, complementing their standard visual audit with in-person interviews and a documentation review.
Rather than rely solely on what they might observe by chance, this approach scrutinizes the facility from three perspectives: what the written procedures say, what key team members report, and what actually takes place during operation. In our experience, this is a much more effective way to identify hidden Annex 1 gaps and address one of the FDA’s most common reasons for issuing an observation: failure to follow SOPs.
After the gap assessment process
The gap assessment process will yield a report identifying your site’s current state of compliance relative to the applicable sections of Annex 1. Depending on the specific approach used by your gap assessment team, states of compliance may range from fully compliant to minor or major out-of-compliance issues, with color-coding or other visual cues in place to identify high-priority areas.
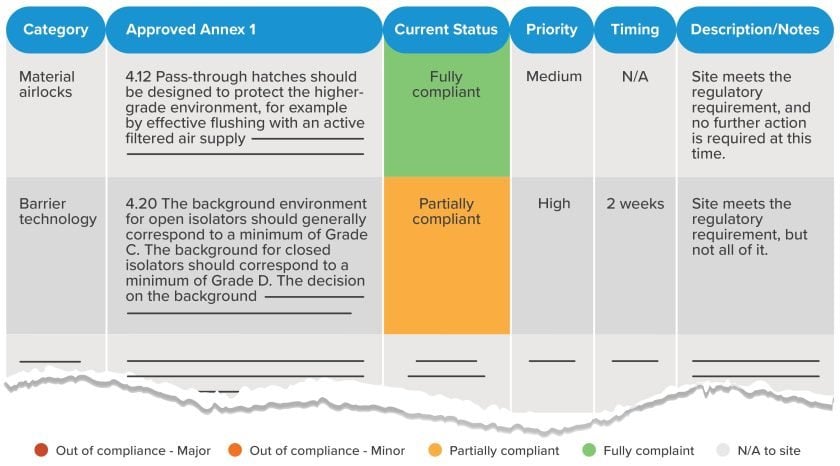
4. What are some common Annex 1 compliance gaps?
Manufacturers should remain vigilant about all potential sources of Annex 1 non-compliance. A few common areas of concern include:
- Process closure
Intervening in a Grade A space through an open door is a thing of the past. Manufacturers must ensure that barrier systems and aseptic handling operations are automated, controlled and closed, and they must maintain Grade A continuity for critical direct and indirect product contact parts from the point of sterilization (i.e. an autoclave) to the point of use. Manual loading operations will be scrutinized more than ever before, particularly where no barrier system is in place.
- Pass-throughs
It will be difficult to justify using simple metal boxes (“passive” pass-throughs) for transferring materials from lower classifications to higher classifications, particularly into Grade B spaces. Pass-throughs must be designed to protect the higher-grade environment, and manufacturers must provide a validatable process for reducing microbial and particulate contamination. This can range from active air supply to the pass-through to VPHP or UV sanitization.
- Single-use systems
Manufacturers must qualify aseptic connectors and prove single-use system closure for each batch. Leachables and extractables must be assessed by product. Don’t forget PUPSIT!
Next Steps Toward Annex 1 Compliance
5. I found Annex 1 gaps in my manufacturing operation. What now?
Identifying gaps in your Annex 1 compliance is one thing. Remediating those gaps—that’s your next challenge.
Prioritize your compliance gaps
The first step is to assign a numerical value to each Annex 1 gap based on urgency and potential impact on patients. This will help you quickly structure and prioritize issues.
Regulators recognize that it may take time to close compliance gaps, and that manufacturers will address those gaps according to criticality. Major issues require immediate action while minor issues may be accommodated on a longer timeline.
Analyze your options
There is often no single, clear solution to address a particular compliance gap. Instead, there are typically a variety of potential avenues for remediation, each with its own advantages and tradeoffs.
Your AEC team can help you evaluate these potential avenues and understand capital cost implications, impacts to ongoing operations, efficiency considerations and more. This expertise will contribute to a strategic options analysis.
Within the options analysis framework, your AEC team can use a variety of tools to evaluate potential solutions, from scaled sketches and comparison charts to help you conceptualize relatively simple changes to sophisticated computational modeling to pressure-test complex, high-budget renovations. The goal is to rank potential solutions based on the criteria that matters most from the perspective of both risk management and business outcomes.
In every case, understanding the bigger Annex 1 picture and examining the impacts of individual compliance solutions on your operation as a whole is key. This is where AEC experience married with holistic regulatory and operational expertise really pays off. Consider, for example, learning from your gap assessment that you need to add PUPSIT capabilities to your sterile operation. That means a significant CapEx investment, particularly in the case of stainless steel systems. But that’s not all: in addition to upfront costs, adding PUPSIT will cost you in terms of processing time—several hours per batch, in most cases. It also means requalifying portions of your filling lines and updating your product qualifications, airflow visualization studies (i.e. smoke studies), aseptic process simulations (i.e. media fills) and regulatory filings.
Analyzing these implications from all angles allows you to choose an appropriate way forward, plan your capital spending accordingly and avoid surprises as you close in on Annex 1 compliance.
“I need an Annex 1 gap assessment and a capital project delivery plan for closing any identified gaps. Who should I have on my team?”
A strong Annex 1 gap assessment and project delivery partner should offer:
- A comprehensive assessment approach
Assessing your operation from just one perspective (such as a visual audit) leaves you vulnerable to unidentified gaps that could impact future regulatory outcomes. Instead, invest the time to understand your operation in terms of documented procedures, stated claims, and actual practices.
- A good understanding of capital costs
After identifying compliance gaps, your next step is to find and analyze options for remediation. This is much easier when working with a gap assessment team that’s embedded within an experienced AEC firm. That AEC partner will understand the relationship between your available capital and the priorities identified in your gap assessment.
- Deep knowledge of current regulations
As a cautionary tale, consider a manufacturer who adds new airlocks in order to address an Annex 1 compliance gap. A perfect solution—until that manufacturer learns that the materials used to finish those airlocks are non-compliant. To avoid scenarios like this, work with experts who offer a holistic understanding of the regulatory landscape and its facility implications.
The bottom line: Annex 1 compliance for sterile or aseptic manufacturers starts with the right expertise
Are you working toward Annex 1 compliance for your sterile or aseptic manufacturing operation, but you’re not sure where your gaps are, what to do about them, or the role of your Contamination Control Strategy?
Reach out to our team of experts. We have regulatory experts with the answers you’re looking for to ensure that your sterile or aseptic manufacturing process is efficient, compliant, and—above all—delivering products that are safe for the patients who depend on them.