
Return to top


ATMP innovation has moved faster than the GMP guidelines that govern it. To bridge that gap and ensure the safety and quality of their products, ATMP manufacturers need a risk-based approach that’s grounded in a deep understanding of their materials, their processes, and the factors that impact their final product.
This is the final installment in our four-part series, “The ATMP Manufacturer’s Guide to Commercialization and cGMP Compliance.” For access to the full series, sign up here.
When a promising new product type enters the life science market, it takes time for manufacturers, regulators, and the industry as a whole to build consensus around the best and safest way forward.
This was true when monoclonal antibodies (mAbs) emerged several decades ago, introducing novel technologies which have since become standard issues. Now ATMPs are the new products in town, characterized by high-speed innovation and business models that look nothing like the large-volume, single-product commercial enterprises that predate them. Consider personalized ATMPs, for example, which bring unique challenges into play:
- Scalability: Each batch is a product for a target population of one. That means manufacturers need to make a high volume of very small product batches, all within the same facility.
- Sterility: Sterile filtration is likely not possible, which means the entire manufacturing lifecycle may need to meet the rigors of sterile and aseptic processing.
- Variability: Because personalized ATMPs depend on starting material collected directly from the patient, variability is a persistent issue.
To address these and many other unique challenges, ATMP manufacturers need to work in dialogue with regulators to develop tailored, risk-based approaches that bring the principles of quality and safety into the ATMP facility, particularly when the rules are not yet fully harmonized and could cause confusion.
But what do we mean by a risk-based approach, and how can ATMP manufacturers use that approach to navigate the continuously evolving landscape of GMP guidance? In this article, we’ll break that question into five parts:
- What are the most relevant GMP guidelines and regulations for ATMPs?
- Are current ATMP guidelines and regulations harmonized?
- Are all ATMPs the same from a regulatory perspective?
- Where is the GMP boundary in an ATMP manufacturing facility?
- What is the best way to set yourself up for regulatory success in the ATMP industry?
Acronyms to know
EC — The European Commission
EMA — The European Medicines Agency
PIC/S — The Pharmaceutical Inspection Co-operation Scheme
FDA — The United States Food and Drug Administration
GMP — Good Manufacturing Practices
RBA — Risk-Based Approach
QRM — Quality Risk Management
1. What are the most relevant GMP regulatory documents for ATMPs?
Until 2017, ATMPs did not have stand-alone GMP guidelines or regulatory documentation. Instead, they sat under the broad umbrella of “biologics.” As the ATMP industry matured, manufacturers and regulators recognized the need to differentiate ATMPs from other biologics and support their development with specific, tailored regulatory guidance.
That broad consensus kicked off a cycle of mutual effort. Today, regulators are at work keeping pace with advances in ATMP manufacturing, and manufacturers are working equally hard to incorporate updated guidance documents and regulations into their processes and facility design.
ATMP guidelines and regulations: A history
The European Commission revised Annex 2 of EudraLex Volume 4: EU guidelines for Good Manufacturing Practice to include specific considerations for ATMPs.
The European Commission published EudraLex Volume 4, Part IV: Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products.
The European Commission updated Annex 2 of EudraLex Volume 4: EU guidelines for Good Manufacturing Practice to clarify that it is not applicable to ATMPs.
PIC/S revised their Guide To Good Manufacturing Practice For Medicinal Products Annexes to split Annex 2 into Annex 2A (Manufacture of advanced therapy medicinal products for human use) and Annex 2B (Manufacture of biological medicinal substances and products for human use).
The European Commission updated their EudraLex Volume 4: Guidelines on Good Manufacturing Practice to include a revised Annex 1: Manufacture of Sterile Medicinal Products. PIC/S followed one year later with a nearly identical update to their Guide To Good Manufacturing Practice For Medicinal Products Annexes.
A short introduction to current GMP guidance documents:
From The European Commission: EudraLex Volume 4, Part IV: Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products
In 2012, when the EC updated their GMP guidelines to include ATMPs for the first time, most ATMP products were still in the early stages of clinical-scale development. Five years later, as the FDA approved a CAR-T cell therapy for the first time, the EC recognized that the maturing ATMP industry needed a dedicated set of standards and best practices within their GMP guidelines. Thus, Part IV emerged. This decision diverged from prevailing opinions that ATMP guidance should fall within biologics, and instead created a new, dedicated category for ATMPs within the GMP guidelines. The Commission later put these updated ATMP guidances into a necessary requirement and simultaneously updated Annex 2 of their GMP documentation to remove all reference to ATMPs. From that point on, ATMP manufacturers were advised to rely solely on the new Part IV.
Part IV draws from established GMP principles while recognizing the specific needs and challenges of ATMPs. It emphasizes the role of a risk-based approach (RBA) and demonstrates (through specific examples) how RBA gives ATMP manufacturers ownership over the control measures required for their unique manufacturing process. Most of its language refers to ATMPs in general, leaving readers to interpret how these guidelines apply to specific ATMPs with unique risks and properties, as we’ll discuss later in this article.
Significantly, Part IV includes a statement that appears to acknowledge the rapid pace of change in the ATMP industry and the value of uninhibited innovation. In section 1.15, the document reads in part: “These Guidelines do not intend to place any restrain on the development of new concepts of new technologies. While this document describes the standard expectations, alternative approaches may be implemented by manufacturers if it is demonstrated that the alternative approach is capable of meeting the same objective.”
It appears that regulators are aiming for an appropriate balance between the quality, safety, efficacy and traceability of ATMPs and the need for flexibility and adaptability as the science of ATMP manufacturing evolves.
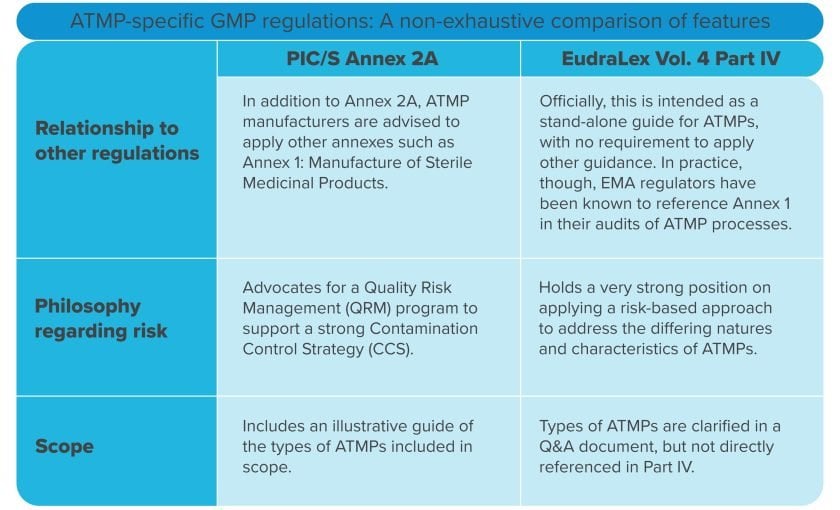
Historically, the PIC/S GMP guide has closely mirrored the European Commission’s GMP guide, but these documents diverge on the topic of ATMPs. Rather than establish a stand-alone regulation like the Commission, PIC/S has chosen to keep ATMPs within its GMP annexes. To accomplish this, it split Annex 2 into two sections. Annex 2A now applies to ATMPs, while Annex 2B covers all other biologics.
From the European Commission and PIC/S: Annex 1 (Manufacture of Sterile Medicinal Products)
Released in 2022, this was the first official Annex 1 update in fourteen years. It is a document with immense weight and influence around the world, and it has far-reaching implications for all life science manufacturers.
Annex 1 applies to process streams that require aseptic handling and sterile conditions. That typically means everything downstream of a sterile filtration step (such as fill-finish operations), although in cases where a process flow cannot be sterile filtered, that boundary typically moves further upstream.
Because they ultimately are an aseptically handled drug product, ATMPs may fall under the purview of Annex 1. However, when the EC released Part IV five years before the official Annex 1 update, they clearly stated that no other Annexes applied to ATMPs. PIC/S adopted a different approach: in their ATMP-specific Annex 2A document from 2022, they frequently reference Annex 1 and define scenarios when manufacturers should operate “in line” with Annex 1 principles.
For manufacturers operating in Europe, these differing points of view could cause confusion. Many choose to apply Annex 1 even if it’s not strictly mandated by Part IV, particularly if they intend to sell their products in areas of the world where PIC/S is a reference for the competent national regulatory authority. This approach invites its own challenge, though: many ATMPs, particularly personalized therapies, cannot undergo sterile filtration, which means moving the Annex 1 boundary. As we’ll discuss later, this can have serious implications for the complexity, cost, and even quality of an ATMP operation.
2. Are current ATMP GMP regulations harmonized?
Regulators and manufacturers are in dialogue, continuously working to expand and harmonize the guidelines that apply to ATMPs. But ATMP research and production isn’t slowing down to wait for regulations to catch up. Manufacturers need practical strategies to make the “as is” regulatory environment work for them right now. That means interpreting the intent behind Part IV, Annex 2A, and Annex 1—particularly in areas where the rules, as written, diverge from one guidance to another.
Differences between regulations could pose a challenge
There are many reasons why ATMP regulators may encounter differences from one guidance or regulatory document and another:
- Regional perspectives: Regulators in one region may establish requirements or leniencies not recognized elsewhere. Manufacturers who plan to operate or sell their products in more than one region must understand and navigate this complexity.
- Timing: Given the pace of innovation in this industry, a lag between the science of ATMPs and the regulations governing them is expected—and that lag will change from agency to agency.
- Different regulatory philosophies: Some contradictions between existing regulations come down to a divergence in perspective. For example, Part IV allows for the use of multiple laminar airflow units in certain low-risk scenarios, while the segregation standard outlined in Annex 1 sets a much stricter expectation.
These differing philosophies can affect if (and how) new technologies are developed. According to Part IV, for example, manufacturers may use closed systems in a room with a Grade D background environment. This incentivises the development of new systems featuring closed technology.
In contrast, the Annex 1 document lays out different criteria for aseptic processing, as well as for scenarios where a system’s integrity is at risk (such as processes that feature single-use technologies). According to these guidelines, manufacturers should locate the closed system in a Grade A environment. A lower classification may be possible from a regulatory perspective only if the manufacturer can demonstrate integrity at every usage (such as through pressure testing and/or monitoring). Both scenarios—maintaining a Grade A environment or proving integrity—impact the development of (automated) single-use kit assembly systems and lower the motivation to innovate by establishing impractical or idealistic provisions.
The solution: A risk-based approach based on the principles behind the regulations
When guidance documents, as written, do not directly address a specific ATMP manufacturing scenario, the solution is to look for the intention behind those documents. This is where a risk-based approach (RBA) is critical.
A robust RBA scientifically identifies the risks inherent to a unique ATMP manufacturing process, based on a holistic understanding of the product you’re making, the materials and equipment required, your process and its degree of closure, and other critical elements.
It’s important to note that establishing a strong RBA doesn’t eliminate all risk. In fact, as manufacturers of almost any product type know, the concept of “zero risk” does not exist. Instead, the goal of the RBA is to demonstrate that your ATMP operation meets the quality, efficacy, and reliability principles behind today’s regulations.
Meeting regulatory expectations via a risk-based approach, rather than by applying written requirements to the letter regardless of their suitability, can challenge both manufacturers and regulators. But facing this challenge will pay off. It will free you to pursue innovation beyond the boundaries of as-is legislation, while helping you communicate effectively with regulators about the scientific rationale behind your risk management approach. You’ll have the flexibility your business needs, the clarity that regulators require, and the quality and safety that patients expect from your products.
3. Are all ATMPs the same from a regulatory perspective?
Early in 2021, the EMA published a Q&A document clarifying the types of ATMPs covered by the EC’s Part IV. Not long after that, PIC/S went further by choosing to include an illustrative guide of ATMP manufacturing activities directly within their new Annex 2A. These were important regulatory milestones: for the first time, the overarching category of “ATMPs” had been atomized into specific products. Gene therapies, somatic cell therapies, and tissue-engineered therapies are included in this scope, as well as ATMPs combined with other therapies or medical devices.
Some regulating authorities go even further, expanding their definition of ATMPs to include other products they deem appropriate. Sub-classifications can differ between regions; European authorities explicitly categorize tissue-engineered products (TEPs) under the ATMP umbrella, for example, while authorities in the US operate a broader classification system that incorporates some TEPs based on specific criteria.
These distinctions between different ATMPs matter, because they allow regulators to recognize that the manufacturing processes, modes of action, and intended indications for use vary from product to product, which in turn lays the groundwork for tailored, quality-based approaches to regulatory compliance.
Annex 1 and the operational complexity of personalized ATMPs
Identifying the considerations in play for different ATMP modalities is a step forward for both regulators and manufacturers. Still, challenges remain—particularly for manufacturers with personalized therapies in their pipeline.
Because each batch is patient-specific, these manufacturers potentially need to make hundreds of individual products within the same facility. From a risk perspective, this puts manufacturers of personalized therapies in the same position as multimodal manufacturers. Campaigning between products within the same suite exposes them to an elevated risk of cross-contamination, and with so many product-specific materials and components in play, there’s also the potential for inadvertent mix-ups.
To address these risks, multimodal manufacturers have learned how to optimally deploy robust control measures, such as automated track-and-trace technologies, strict segregation strategies, and extensive automation and process closure.
To use these measures efficiently and effectively, the first step is an evidence-based QRM program designed to identify potential vulnerabilities. In many cases, the authoritative text to help manufacturers address these vulnerabilities is Annex 1, which emphasizes the role of strict contamination control to maintain the quality and safety of sterile products—a category into which personalized therapies squarely fit. However, the size or sensitivity of the living cells in many personalized therapies means they can’t undergo a sterile filtration step, which expands the scope of Annex 1.
However, simply applying Annex 1 to the whole manufacturing process is often not feasible from a cost and flexibility perspective, nor is it necessarily beneficial in terms of product integrity. In fact, adhering to the entirety of the Annex 1 document during upstream processing may—in some cases—work against the quality and safety intentions of the regulation.
For example, Annex 1 advocates for the use of isolators and Reduced Access Barrier Systems (RABs) to preserve an aseptic processing environment. But these technologies are not always the solution for ATMP manufacturers, who may need to rely on technological adaptations and robust closed systems to ensure aseptic handling.
From a material handling perspective, using an isolator for upstream processing could introduce significant challenges for ATMP manufacturers. The bulky gloves necessary for working inside an isolator, for example, could limit an operator’s ability to undertake certain quality measures. Thinner glove material may help, but that introduces its own challenges, such as the risk of a tear, putting both the operator and the product at risk.
Cleaning and disinfecting upstream isolators may also be a challenge, particularly in cases where automated decontamination via vaporized hydrogen peroxide isn’t possible because of the delicate nature of personalized therapies. A manual wipe-down may be the only alternative—a difficult process to validate against the rigorous standards of Annex 1.
As always, a deep understanding of the quality risks associated with your unique process is the key to applying Annex 1 and other relevant guidance documents appropriately, ensuring that your personalized ATMP manufacturing operation is safe, efficient, and scalable.
4. Where is the GMP boundary in an ATMP manufacturing facility?
GMP guidelines establish standards and best practices for drug manufacturers. For those manufacturing traditional biologics, the production process is typically easy to divide along a clear GMP boundary; everything downstream of that boundary should follow GMP guidelines in order to ensure the safety and quality of finished products.
For ATMP manufacturers, that boundary may not be as clear. ATMP manufacturing often relies on starting materials such as plasmid DNA or viral vectors, which have their own complex manufacturing processes. Given their impact on the quality, safety, and purity of the final product, these processes may need to follow GMP standards, as well.
From this perspective, the GMP boundary for ATMPs could arguably extend into the supply chain and through hundreds of operations across multiple sites.
This broad-scale approach is not necessarily the answer. Applying the same level of GMP stringency across the entire process may not add value from a quality perspective, and it could negatively impact the speed, cost, and accessibility of life-saving therapies. Instead, manufacturers should apply a QRM program to design a progressively more stringent GMP approach. In other words, as the risk to product quality and safety increases, so should the level of GMP control.
The 2021 publication of Annex 2A from PIC/S helped to clarify this point (the EMA released a related Questions and Answers document soon after). In Annex 2A, PIC/S illustrates the distinction from upstream manufacturing steps, where manufacturers should use a QRM approach to determine an appropriate level of GMP application at different stages in the manufacturing process.

5. What is the best way to set yourself up for regulatory success in the ATMP industry?
Early engagement with regulators can help manufacturers navigate the complex and sometimes contradictory framework of rules, guidance documents, and overarching principles that apply to ATMP manufacturing.
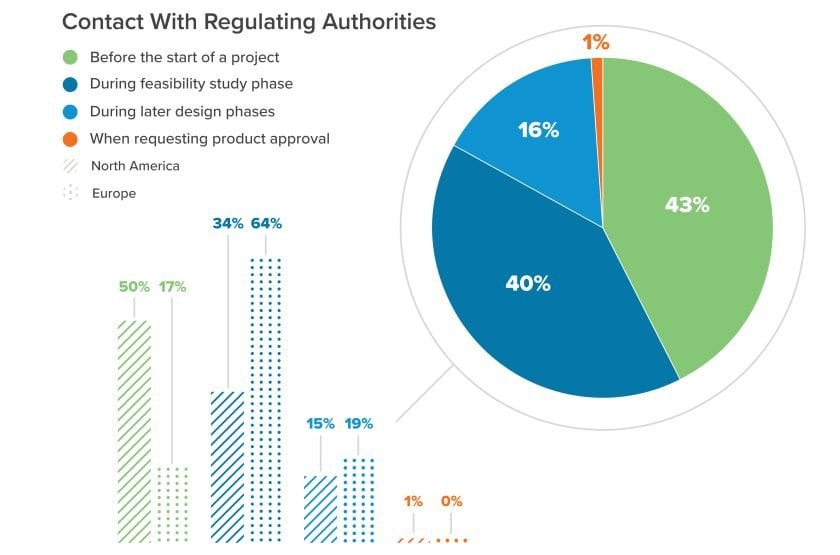
When we surveyed nearly 500 companies for last year’s Horizons: Life Sciences report, we noted a few different philosophies in play when it comes to regulatory engagement. The majority of manufacturers contact regulators during the feasibility study phase, but a meaningful number are starting to work with regulators before a project even begins. That’s an encouraging sign; the earlier manufacturers make that initial contact, the better prepared they’ll be to align early design decisions with regulatory requirements.
Stand guard over quality—without standing still
ATMPs are characterized by change. Manufacturing approaches, supporting technologies, regulatory guidance—they’re all evolving, and fast. To keep up, you need a manufacturing approach that’s both robust and flexible, and a strategy for controlling and mitigating quality risks that’s tailored to your situation and scientifically defensible to regulators. But how can you get there, when existing GMP guidelines are frequently evolving?
A risk-based approach is the answer. With a deep understanding of what your materials are, how your process works, and where you need to apply stringent control measures to protect the patients who rely on you, you have the insight you need to apply the principles behind regulatory guidance, even when the guidance itself falls short of your situation.
To develop and validate your risk-based approach, lean on the experience of people who know the ATMP world, including design experts, process engineers, and regulators themselves. With this team in place and a risk-based approach guiding you forward, you’ll have what you need to develop a commercial-scale ATMP operation that’s continuously innovating and delivering the high-quality products that patients need, now and far into the future.
It will take time and ongoing collaboration to arrive at a set of comprehensive, responsive, and fit-for-purpose ATMP regulations—and even then, unanticipated technologies, processes, and drug types will continuously emerge, necessitating further updates from regulators and a flexible, risk-based approach from manufacturers.
Frequently Asked Questions: GMP guidelines for ATMP manufacturing process
This article provides a practical overview of how to apply GMP guidelines for ATMP manufacturing by using a risk-based approach. It outlines the key GMP frameworks (EudraLex Vol 4 Part IV, PIC/S Annex 2A, EU Annex 1), explains how they apply to advanced therapy medicinal products (ATMPs), and clarifies how manufacturers can define GMP boundaries, ensure compliance, and maintain product quality across the entire ATMP manufacturing process.
GMP guidelines for ATMPs are designed to reflect the unique risks and production models of cell and gene therapies. Unlike traditional biologics, ATMP GMP frameworks allow flexibility in manufacturing controls when supported by scientific risk assessments. These guidelines emphasise quality risk management (QRM), aseptic processing, and the control of starting materials and raw inputs that may directly impact product safety.
A risk-based approach allows manufacturers to apply GMP controls proportionally to identified product and process risks, rather than following a one-size-fits-all model. This method ensures regulatory compliance while supporting innovation, scalability, and patient safety — critical factors for ATMP manufacturing where every batch and patient profile may vary significantly.
The GMP boundary includes all steps that directly influence product quality — from donor or cell collection through final formulation and release. In the ATMP manufacturing process, upstream activities such as material sourcing or vector preparation may also fall under GMP control if they pose a direct impact on product safety or consistency. Clearly defining this boundary is key to efficient, compliant production.
Manufacturers should engage with regulatory authorities early, document their risk-based rationale, and design flexible, quality-driven facilities that can adapt to evolving GMP expectations. Combining robust QRM practices with transparent data, traceability, and well-defined manufacturing boundaries builds confidence in compliance and accelerates regulatory approval.


An introduction to ATMP manufacturingHow the ATMP and cell and gene therapy industries can prepare to scale manufacturing operations safely, sustainably, and strategically.
Read More
How the ATMP and cell and gene therapy industries can prepare to scale manufacturing operations safely, sustainably, and strategically.
Read More