Every patient who receives a drug injection trusts that it’s safe. That trust depends on robust contamination control strategies built into the manufacturing process. Nowhere is this more important than during sterile filtration—the final step before drug products are filled into vials, syringes, or cartridges.
For filtration to work as intended, manufacturers rely on the integrity of both the filtration process and the filters themselves. Post-use integrity testing, performed as a requirement for product release, has long served as a cornerstone of sterile manufacturing, confirming the condition of the filter at the end of filtration but not necessarily that it remained integral throughout the process. To address this risk, Annex 1 places increased emphasis on Pre-Use Post Sterilization Integrity Testing, or PUPSIT. While not a new concept, PUPSIT became a clear regulatory expectation with the 2022 revision of Annex 1. By helping reduce the risk of filter failures going undetected, particularly in cases where product or process conditions may mask defects, PUPSIT strengthens assurance of product sterility beyond post-use testing alone.
By adding another layer of protection against product contamination, PUPSIT could protect patients from harm and prevent batch loss. Implementing PUPSIT can be challenging, particularly for manufacturers with existing processes in place. It requires time and capital investment, as well as careful alignment with regulatory expectations. A strong understanding of PUPSIT’s technical and regulatory requirements can help minimize these challenges. The following five questions examine the fundamentals of PUPSIT and key considerations for designing a robust, PUPSIT-capable sterile filtration process.
1. What is PUPSIT?
Pre-Use Post Sterilization Integrity Testing (PUPSIT) is a process step designed to detect flaws or defects in sterilizing-grade filters prior to use. It is generally performed after filtration systems have been fully assembled and deemed operational for the filtration of bulk drug product.
To understand PUPSIT, it helps to examine the role of filtration in the aseptic and sterile drug product manufacturing process. Manufacturers must pass bulk drug products through a sterilizing-grade filter prior to the filling step. These filters are required to have a pore size of at least 0.2 microns, as this size is effective at retaining common bacterial contaminants, most notably Brevundimonas diminuta (~0.3 micron), the standard test organism used in both the US and European Pharmacopoeia. While these filters are primarily designed for microbial retention, they also remove particulate at the sub-micron level.
The processes required to sterilize these filters before use typically involve aggressive methods like steam-in-place (SIP) or gamma irradiation. These methods could compromise the filter’s integrity, resulting in cracks or pinholes which put the quality of sterilized product at risk and cause manufacturers to lose part or all of a batch and the associated potential revenue. Product loss isn’t the only risk of a damaged filter, though. If downstream integrity testing fails to detect a problem, patients could be exposed to non-sterile product.
A phenomenon called “flaw masking” makes this risk a real possibility. Flaw masking occurs when the sterilization or assembly process damages the filter in a way that allows bacterial penetration, but that flaw is “masked” or plugged during the filtration process. As a result, the filter may generate a false pass during post-use integrity testing. This concept was introduced and studied by the Sterilizing Filtration Quality Risk Management (SFQRM) consortium, a collaborative initiative involving the Parenteral Drug Association (PDA) and BioPhorum. The article linked below documents how this was demonstrated with OvaltineTM (yes, that Ovaltine).
The PUPSIT requirement was developed to address this risk of flaw masking caused by fouling during the filtration process, allowing a damaged filter to pass post-use integrity testing.
The Ovaltine experiment: How a breakfast drink shaped the filter integrity conversation
Driven by anecdotal evidence of flaw masking, PDA and BioPhorum’s SFQRM formed a task group to study the potential for masking through a robust trial. They needed a low-cost fluid to stand in for a drug substance—one that would behave like a biologic with a high foulant concentration. That’s where Ovaltine came in.
As a result of this masking study, the task group found that flaw masking is possible “under extreme circumstances of fouling and blocking of a sterilizing grade filter.” This conclusion helped build regulatory consensus in the EU, where pre-use integrity testing is now broadly expected.
2. What are the regulatory expectations for PUPSIT?
Even though the term PUPSIT wasn’t explicitly used in previous versions of Annex 1: Manufacture of Sterile Medicinal Products, regulators nonetheless recommended pre- and post-use testing. But it wasn’t until drafts of the newest Annex 1 revision were circulated for public consultation in 2017 that PUPSIT appeared as a formal requirement for the first time.
The revised Annex 1, published in 2022, includes the following language in paragraph 8.87:

Annex 1 also provides specific guidance for filter sterilization, including understanding potential causes of filter damage and maintaining a holistic view of supply chain audits.
Most updated Annex 1 guidelines, including this PUPSIT requirement, became enforceable across all 27 national competent authorities of the EU in August 2023. The Pharmaceutical Inspection Co-operation Scheme (PIC/S) has also brought the updated Annex 1 guidelines to all 54 of its regulatory body members and harmonized with the PUPSIT requirement. Annex 1 applies not only within member countries but also to any country shipping aseptic and sterile products into these markets.
In the US, FDA regulators are broadly aligned with the principles of Annex 1. While PUPSIT is not necessarily a strict requirement for products sold in the US market, the FDA supports a risk-based approach to confirming the integrity of the filtration step. Manufacturers who responded to a recent industry survey conducted by BioPhorum’s SFQRM team reinforce this impression, telling surveyors that the FDA seems to be taking cues from EU Annex 1 during inspections. In other words, to position themselves for a successful regulatory inspection, US-based manufacturers should demonstrate that they’ve proactively risk assessed the requirement for PUPSIT as part of their overall contamination control strategy.
To meet this regulatory expectation, manufacturers should adopt a rigorous risk-based approach guided by quality risk management principles. The goal of this approach is to identify specific product- and process-related risks and define appropriate mitigation strategies. The resulting risk assessment documentation will demonstrate to regulators that manufacturers have thoroughly evaluated and addressed the potential for flaw masking, and that they’ve implemented PUPSIT where appropriate.
In rare scenarios, manufacturers may determine that the complexity and risks of implementing PUPSIT outweigh its potential benefits. The volume of bulk drug product may also influence this decision. Manufacturers processing small volumes, for example, may have a case for not implementing PUPSIT, though it’s important to note that there is no fixed definition of ‘small’.
Manufacturers who choose this alternative direction must defend their position to regulators using science-based, data-driven evidence, explaining how they intend to ensure product sterility and patient safety through strong process controls and other mitigation strategies. In other words, difficulty to implement the process alone is not considered sufficient rationale for omitting PUPSIT.
A risk index matrix that evaluates the likelihood and severity of potential failures can help justify the decision. Relevant risk factors that may include:
| Process attribute (why it matters) | Low risk indicators | Medium risk indicators | High risk indicators |
| Regulatory/regional expectations | Local regulators allow flexibility and justification is strong | Mixed regulatory expectations; guidance recommends a risk-based approach | Regulatory guidance (such as EU Annex 1) expects verification unless justified otherwise |
| Product fouling potential (masking risk) | Low solids, low particulates, low risk to mask defects | Occasional particulates or moderate protein loads | High particulate load, viscous or foaming fluid that can clog/mask marginal defects |
| Historical deviations/supplier complaints | No prior filter failures, stable history | Occasional deviations observed but adequately investigated | Recent filter integrity failures, unexplained sterility incidents |
| Sterilization method & robustness (gamma, steam, etc.) | Sterilization proven within the validated filter design space; process is well-controlled | Occasional excursions/ borderline cycles | Sterilization method known to stress filter; poorly controlled cycles |
| Filter configuration & redundancy (single vs. redundant filters) | Validated redundant filtration with independent paths | Redundancy present but components/valves are shared | Single filter for entire batch (no redundancy) |
| Single-use assembly vs. reusable housings | Single-use sealed, gamma-irradiated assemblies with validated packaging | Single-use but assembled on site; some open steps | Reusable housings requiring on-site assembly/sterilization |
| Manual valve manipulation & intervention risk | Fully welded/closed system and automated valve operations | Some manual interventions but controlled | Multiple sterile-side valve operations and complex manipulations |
3. What tests are performed during PUPSIT?
The PUPSIT process typically features one of two commonly used filter integrity tests: the bubble point test and the forward flow or diffusion test. Manufacturers must consider factors such as filter size and testing sensitivity requirements to select the appropriate test for their scenario, though both tests follow the same basic approach.
- Saturate the filter with a wetting fluid
- Perform the integrity test using a compressed gas and integrity testing equipment
- Flush the filter assembly to remove residual wetting fluid
Bubble point test
Relative to the diffusion test, the bubble point test involves a simpler process that’s suitable for detecting the largest pore sizes on smaller filters. It works by measuring the pressure required to overcome the surface tension of the wetting fluid and push it through the filter membrane’s largest pores, causing bubbles to appear. If this “bubble point” matches vendor-supplied specifications when executed with vendor-specified fluid (usually water), the filter assembly passes. If product is used as the wetting medium, the manufacturer must determine and validate a new product-specific bubble point.
Forward flow or diffusion test
This test, which is more complicated to perform, is suitable for larger filters where very small defects may be possible, requiring heightened testing sensitivity. To run this test, manufacturers apply gas through the wetted filter at a constant pressure set below the bubble point. The rate of gas flow through the membrane determines filter integrity.
Wetting fluid used in PUPSIT: An important consideration
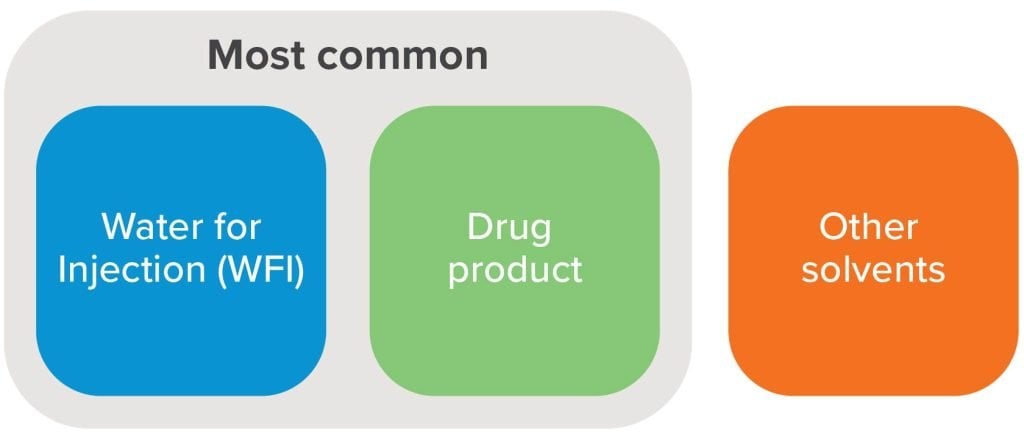
The first step in the PUPSIT process is to precondition the filter membrane by saturating it with a wetting fluid. Manufacturers generally have three main options, each with its own advantages and trade-offs. Selecting the appropriate fluid requires a thorough assessment of process requirements.
Water for injection (WFI)
Though it’s the least costly of these three options and widely available in manufacturing facilities, WFI requires additional process steps to flush residual water from the product flow path to minimize the risk of product dilution. In some cases, WFI may not be compatible due to the use of hydrophobic filter membranes, such as polytetrafluoroethylene (PTFE), which do not readily wet with water.
Drug product
Using drug product as the wetting media allows for testing under the exact conditions in which filtration will occur and gives the most representative integrity test. This is also a suitable option in cases that involve hydrophobic filter membranes. But manufacturers who rely on bulk drug product as their wetting media must carefully consider the volume necessary to fully saturate the filter, especially given the potential for product waste.
Other solvents
Though ethanol and other flammable solvents are common in post–use integrity testing, they are not often used for PUPSIT. When utilizing ethanol, isopropanol, or other solvents for filter testing, manufacturers must consider safety factors that have far-reaching implications for both process and facility design, as governed by the International Building Code (IBC), the National Fire Protection Association (NFPA) and the International Fire Code (IFC).
For example, the area and equipment involved may require electrical classification due to open or closed processing of flammable or combustible compounds. Building codes also limit the maximum quantities of flammable solvents allowed in a control area, based on factors such as whether they’re handled in open or closed systems. Before incorporating flammable solvents into a PUPSIT process, manufacturers should evaluate these requirements from an overall control area perspective, with consideration for the total volume of flammable solvents used throughout the manufacturing process, including PUPSIT.
4. Which filters require PUPSIT, and how does system design influence implementation?
A filtration system’s design and the number of filters in its flow pathway may impact a manufacturer’s PUPSIT implementation strategy.
Single, double, or triple filter setups: PUPSIT for different configurations
Every filter assembly requires at least one sterilizing-grade filter. Beyond that shared requirement, filtration pathways may diverge in terms of configuration and complexity. Some feature a relatively simple single-filter setup, while others involve redundant filters, additional valves and other ancillary components.
To decide which filters require PUPSIT, manufacturers should consider the following:
Single-filter setup
In this scenario, the single sterilizing-grade filter must be integrity tested both pre- and post-use.
Two-filter setup
When the filtration pathway features two filters, the first is generally considered a conditioning filter, while the second, further downstream, is regarded as the sterilizing-grade filter. Typically, applying PUPSIT on the final filter is considered sufficient. The conditioning filter may require pre-use testing if the risk assessment process reveals an elevated likelihood of fouling or damage from the filter sterilization process.
Three-filter setup
In more complex systems involving three filters, the first filter is not generally tested prior to use. The decision to apply PUPSIT to the second or third filter—or both—depends on fouling risk, process complexity, and other factors identified during the risk assessment process.
Single-use versus stainless steel: Considerations for implementation
The choice between single-use and stainless steel systems affects components, capital expenditures, operational expenditures and overall operations across the entire manufacturing lifecycle, from bioreactors to fill-finish equipment. Sterile filtration assemblies are no exception, with important CapEx and OpEx considerations alongside trade-offs in flexibility, process complexity, sterility assurance, material and sterilization compatibility and other factors.
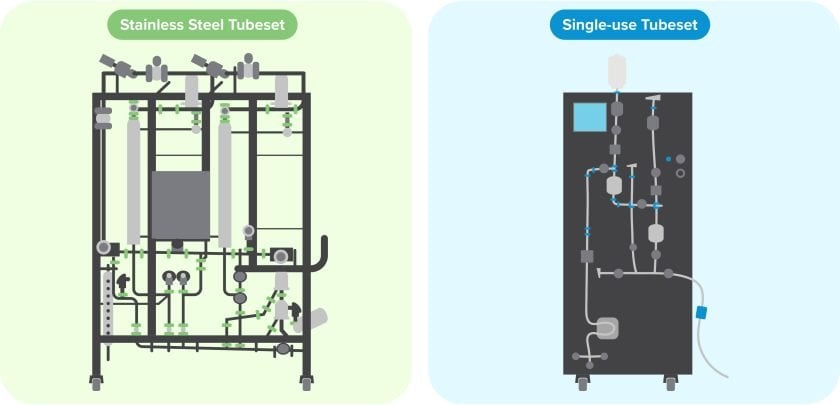
Stainless steel filtration assemblies
Designing and fabricating a stainless steel filtration skid is a complex process. Vendors must ensure that all sloping, venting and other critical features are engineered to support a reliable sterilization flow path. Manufacturers could also consider whether to sterilize filters in an autoclave and then aseptically install them in the filtration assembly, or sterilize them in place as part of a fully integrated skid.
To test filter integrity in a stainless steel skid, operators must configure a flow path to isolate that filter and execute PUPSIT using appropriate wetting, venting and blowdown steps. Drainability is a key factor in stainless skid design. Residual wetting fluids must be completely removed from the filtration assembly to prevent test fluid from impacting product quality. When using pump-around loops, manufacturers must take extra care. These systems can complicate integrity testing by masking defects or creating pressure and flow transients that invalidate results, and they require additional measures to maintain downstream sterility while isolating and testing the filter.
Single-use filtration assemblies
Relying on presterilized, ready-to-use single-use assemblies eliminates the need for SIP or autoclave sterilization, but it introduces other complexities and risks, including supply chain dynamics.
From a PUPSIT perspective, manufacturers can perform the wetting, venting and blowdown steps manually or with automated pinch valves controlled by an execution recipe on a single-use system. In either case, operators must ensure that the pre-sterilized tubing sets are installed and configured correctly so that integrity testing can proceed without breaching the sterile boundary. Similarly, pump-around loops demand particular care, as these configurations increase the risk of operator error during manual steps.
5. What are the key challenges of implementing PUPSIT?
Additional process complexity
Sterile filtration is a complex step in the manufacturing lifecycle for sterile and aseptic products. Adding a pre-use integrity test increases this complexity by introducing new equipment, process steps and training requirements.
For example, PUPSIT often requires additional sterile test assemblies, sterile vents, manifolds with additional filters to maintain the sterile boundary, flush bags and more. This increases both the setup time and the overall complexity of the filtration pathway. Because the process requires manipulation on the sterile side of the filter, there’s a risk of compromising the sterile boundary and introducing contamination. It’s a procedure with the potential to both help and harm, making operator skill and strict adherence to SOPs critical. Poor technique or deviations (such as improper connections or lapses in aseptic practice) can negate the intended benefits.
To reduce these implementation risks, manufacturers can design systems that require minimal handling (through features such as closed/pre-sterile test fixtures or built-in ports), use fully validated integrity testing methods for the product and assembly, implement clear SOPs and competency training and maintain strong control over suppliers and materials.
Compliance risks
Though all manufacturers should begin with a thorough risk assessment, the pathway to regulatory approval may vary depending on the specific scenario. While regulators generally expect PUPSIT for new products and processes, manufacturers with existing products and legacy production lines may be able to defend a data-driven decision to use alternative risk-mitigation strategies instead. However, taking this position without a very strong justification and risk assessment may put compliance at risk, as noted above in Question 2.
Generally, manufacturers who do not currently perform PUPSIT are expected to establish a long-term plan for implementing it in the future, where possible.
Adjacent regulatory requirements
While PUPSIT is a major area of focus, it falls within a larger regulatory context that encompasses filtration process characterization and monitoring. Regulators expect manufacturers to define and monitor filtration parameters established during validation, such as minimum time taken to filter a known volume of bulk solution, pressure differential monitoring during filtration and other important elements of system performance monitoring.
Section 8 of Annex 1, Filter sterilization of products which cannot be sterilized in their final container, outlines many of these interconnected requirements and expectations. Regulators expect manufacturers to demonstrate a clear understanding of their filtration process. For example, manufacturers must either measure temperature or provide robust studies showing why temperature is irrelevant to their filtration step—otherwise, regulators could raise questions. Similarly, if a manufacturer measures differential pressure across the filtration step, or even across each filter, they must have supporting studies to show that these measurements are relevant to the process. “Because we’ve always done it that way” is not sufficient justification.
Evaluating and addressing each parameter as part of a broader regulatory readiness plan is a challenging task that demands experience, careful planning and a holistic strategy for contamination control.
Where to turn for expert PUPSIT guidance and support
Start by reviewing the strategy roadmap for the implementation of a risk-based approach to PUPSIT from BioPhorum’s SFQRM team, written to help manufacturers “use scientific data and risk-based approaches to make and defend decisions” related to PUPSIT.
Manufacturers may also benefit from engaging with an experienced third-party partner who can oversee the risk assessment process, helping manufacturers assess how a PUPSIT implementation will affect process design, facility layout and regulatory compliance. The right partner will also advise on pragmatic details such as testing configurations, skid modifications and PUPSIT training strategies to ensure operator readiness.
PUPSIT is a complex undertaking, and its requirements extend beyond filter integrity testing alone. Now that PUPSIT is a part of Annex 1 compliance expectations, many companies are working to understand its implications for current and future manufacturing. With sufficient data and appropriate risk assessments, choosing not to implement PUPSIT remains an option. However, most manufacturers are advised that any new aseptic sterilizing filtration process will be required to include PUPSIT, unless justified through approved risk assessments.
As with all regulatory situations, early dialogue with regulators is critical to ensure a streamlined approval process. If you’re a manufacturer navigating the decision-making and documentation steps required for PUPSIT, reach out to our team. With our experience in PUPSIT implementation, we can help you chart a clear path to compliance.
Frequently Asked Questions
PUPSIT is a pre-use integrity test performed after filter sterilization to confirm the filter is intact before product filtration. It is required to reduce the risk of undetected filter damage that could compromise product sterility, particularly under EU GMP Annex 1.
PUPSIT detects filter defects before product contact, preventing damage from being masked by fouling during filtration. This ensures that defects caused by sterilization or assembly are identified before they can impact sterility.
PUPSIT is a regulatory expectation under the 2022 revision of EU GMP Annex 1 unless a documented, science-based risk assessment justifies its omission. Cost or implementation difficulty alone is not considered sufficient justification.
The FDA does not explicitly require PUPSIT but expects manufacturers to apply a risk-based approach aligned with Annex 1 principles. Inspectors increasingly look for evidence that the risk of flaw masking has been assessed and controlled.
Key challenges include added process complexity, managing sterile-side manipulations, and ensuring operators are properly trained. Effective system design, validated testing methods, and clear SOPs are critical to successful implementation.