


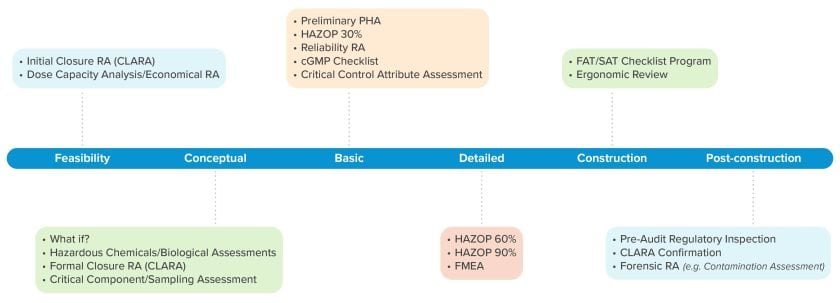

To offer speed to clinic, flexibility, and maximized manufacturing availability, our team conducted a comprehensive process closure analysis risk assessment (CLARA) to inform the design of this facility.
Read More
Return to top
Effective risk assessments provide manufacturers with a systematic, scientifically driven framework for making the right decisions at the right time to support successful outcomes. Regulators expect manufacturers to include risk assessments in their Contamination Control Strategy (CCS) and Quality Risk Management (QRM) programs.
Sometimes viewed as limiters of innovation, risk assessments are, in fact, the opposite. For manufacturers facing time and cost pressures while conceptualizing the facilities and processes of tomorrow, risk assessments are the key to getting new concepts right the first time and ensuring rapid, responsible, and reliable outcomes from one end of the project delivery lifecycle to the other.
Risk assessments comprise a constantly evolving landscape of strategies, from well-established tools such as Failure Modes and Effects Analysis (FMEA) to emerging methodologies like Closure Analysis Risk Assessment (CLARA). Designed to support the implementation of closed systems within the rigors of a strong contamination control strategy, CLARA is a good example of how a systematic risk assessment can help manufacturers meet quality and safety objectives while implementing new strategies that will lead to higher efficiencies, lower costs, and better sustainability outcomes.
- When should manufacturers undertake a risk assessment?
- How do risk assessments benefit manufacturers in the life science industry?
- How should manufacturers prioritize risks identified in a risk assessment?
- How are risk assessments evolving to address new manufacturing strategies and contamination challenges?
- What goes into a successful risk assessment?
Risk assessments: an overview
In the context of life science manufacturing, risk is commonly a function of two key factors:
- Likelihood: The likelihood (or probability) that a risk or hazard will occur.
- Severity: The severity (or impact) of the hazard (on the facility, the project, operators, patients, etc.).
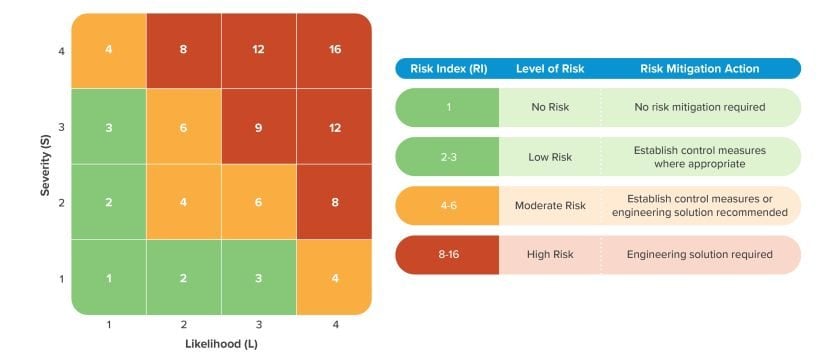
A risk assessment applies pre-established risk criteria to quantify each of these factors independently. By creating a matrix of likelihood and severity, teams can define the risk index of a particular hazard. The risk index steers manufacturers toward scientifically driven conclusions and away from gut feelings or “what worked before.” Manufacturers should incorporate risk criteria into their corporate standards and systematically apply them to all subsequent risk assessments.
Risk Index (RI)
A Risk Index (aka risk ranking) is the result of the function of likelihood and severity (RI = ƒ(likelihood, severity)) of the hazard or risk element. The RI does not include detectability.
For an in-depth look at how to define and prioritize hazards, see section #3 in this article (How should manufacturers prioritize risks identified in a risk assessment?).
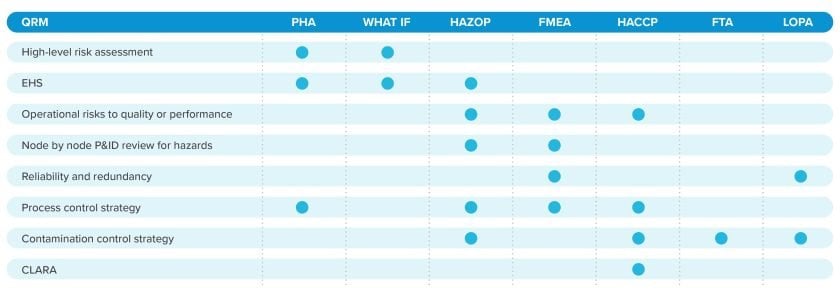
Risk assessment tools and platforms: a sample list
Life science manufacturers have many risk assessment tools at their disposal, each calibrated to support unique objectives at different points in the project delivery or manufacturing lifecycle.
A review of a process or facility early in its development, with the goal of identifying potential hazards, assessing associated risks, and developing related safety measures.
A brainstorming approach where subject matter experts (SMEs) pose hypothetical scenarios to identify potential hazards and assess their consequences. This flexible and creative method is particularly useful in early-stage design.
A structured and comprehensive platform that uses key prompts to identify potential hazards and operational risks. Risk assessment teams can use HAZOP to recognize potential issues by analyzing deviations from the design intent, ensuring safer and more efficient operations.
A proactive tool that identifies potential failure modes in a process or system and assesses their impact on operations. It is typically applied at the Piping and Instrumentation Diagram (P&ID) level when systems are well defined. By ranking the severity, occurrence, and detectability of each failure, FMEA enables biopharmaceutical manufacturers to prioritize risks and implement preventive measures.
Fault Tree Analysis is a top-down, deductive analysis method that identifies the potential causes of system failures. By constructing a fault tree, risk assessment teams can visualize the logical relationships between different failures and pinpoint their root causes. This method is particularly useful in forensic evaluations, which require teams to understand the interplay of various factors to effectively manage future risk.
This semi-quantitative risk assessment technique is designed to evaluate multiple layers of protection, helping to determine their effectiveness in preventing or mitigating hazardous events. By analyzing each layer’s contribution, LOPA helps risk assessment teams ensure that all safeguards are appropriate and sufficient.
This tool helps risk assessment teams identify and control potential problems before they occur. It is designed to mitigate the risk of the product to patients and serves as the platform used in Closure Analysis Risk Assessment (CLARA). It’s based on seven scientific and technical principles:
Principle 1: Conduct a hazard analysis.
Principle 2: Identify critical control points.
Principle 3: Establish critical limits for each critical control point.
Principle 4: Establish critical control point monitoring requirements.
Principle 5: Establish corrective actions.
Principle 6: Establish record-keeping procedures.
Principle 7: Establish procedures to verify that the HACCP system is working as intended.
1. When should manufacturers undertake a risk assessment?
Manufacturers can choose from an arsenal of recommended risk assessment platforms to execute confidently and efficiently at different phases across a project lifecycle, from early feasibility planning through post-construction validation, verification, and improvement.
For example, applying a HAZOP review at 30%, 60%, and 90% project completion gives design teams the opportunity to refine their plans iteratively, minimizing the risk of costly rework or delays caused by late-stage course corrections.
To determine which risk assessment tools are appropriate at different phases across the project lifecycle, manufacturers must consider the type of risk assessment required by their QRM program. Each risk assessment type leverages specific tools to achieve its objectives, helping establish a robust and holistic understanding of the end-to-end drug manufacturing operation.
Risk assessment types: A sample list
This risk assessment is becoming ubiquitous in biopharmaceutical projects. It can serve as an integral part of the Basis of Design for facilities and processes. It focuses exclusively on product and patient safety, evaluating points in a process that are potentially at risk of contamination from the environment. For a closer look at CLARA, see section #4 in this article (How are risk assessments evolving to address new manufacturing strategies and contamination challenges?).
A robust contamination control strategy is now a de facto regulatory requirement in drug substance and drug product manufacturing. The contamination control risk assessment is a tool used to evaluate potential sources of contamination in a drug manufacturing process and provide recommendations for effective mitigation of these risks and a basis for the CCS.
Unexpected system failure and process shutdown are extremely costly to drug substance manufacturers. The cost of unnecessary redundancy in manufacturing and support systems and services is also significant. The reliability risk assessment provides a foundation to predict when and where system and component failures will occur and provide recommendations for appropriate levels of redundancy or alternative risk mitigation measures.
For drug manufacturers, patient safety is critical. However, the safety of the operators and the environment are also paramount. Formal risk assessments addressing the health and safety of operators and the environment are an absolute requirement.
Platform, tool, or type?
A risk assessment platform or tool provides the structure and methodology for conducting risk assessments. Examples include PHAs, HAZOPs, and FMEAs.
A risk assessment type defines the risk assessment’s objectives. Examples include CLARAs, EHS assessments, and reliability/redundancy assessments.

The answers to these vital questions potentially leave facilities, equipment, processes, products, operators, and patients vulnerable to considerable risk. Relying on instincts and guesswork to identify, assess, and resolve the potential impact of these hazards is inappropriate. Systematic risk assessments are the solution.
Within the framework of a QRM program, systematic risk assessments help manufacturers use structured calculations to rank potential hazards, evaluate current safeguards, and document proposed risk mitigations. It’s the difference between predicting the weather by looking at the sky or relying on a detailed, data-driven forecast based on meteorological science—only with much higher stakes involved. Using the right risk assessment tools at the right time, manufacturers can make fact-based decisions on comprehensive and quantitative systems. The potential benefits are numerous:
Improved decision-making
The insights gained from a risk assessment support better, more confident decision-making based on potential risks and their consequences. Management teams can use these insights to allocate resources more effectively while prioritizing safety initiatives.
Improved patient safety
Risk assessment tools provide the groundwork for confident design and process development, ensuring that the products released to patients are safe, high-quality, and reliable.
A strong regulatory strategy
Risk assessments help manufacturers identify and resolve potential observations before they become compliance issues. They demonstrate a proactive and transparent risk-based design approach that anticipates requirements from agencies such as the European Medicines Agency (EMA) or, in the U.S., the Food and Drug Administration (FDA), the Occupational Safety and Health Administration (OSHA), the Environmental Protection Agency (EPA), fire marshals, and others.
Less unplanned downtime and greater operational efficiency
By identifying and mitigating potential risks, PHAs reduce the likelihood of process interruptions. This leads to smoother operations, minimized downtime, and, ultimately, cost savings for the organization. Risk assessments can help manufacturers maximize their uptime by identifying systems prone to issues, establishing predictive maintenance schedules, and informing risk-based redundancy strategies.
Lower capital and operational spending
Applied early in project design, a risk mitigation strategy can help manufacturers simplify their facilities and avoid costly rework, leading to reduced capital spending. Once operational, a facility guided by ongoing risk assessment practices will help manufacturers control costs by finding and addressing inefficiencies, eliminating waste, optimizing energy use, and developing safer, more streamlined workflows to enhance operator productivity.
A new way of designing facilities
Until recently, facility design relied in part on “legacy thinking”: if it worked in previous facilities, it would work again—or so the logic went. But today’s regulators expect more. Manufacturers must establish a Basis of Design that’s grounded in scientific logic, not precedent. A sophisticated QRM must drive all decision-making, ensuring that new facilities are designed to minimize hazards and protect patients and operators. To achieve this and enable a future driven by innovation, not limited by established templates, a risk assessment strategy is key.
Improved sustainability outcomes
The one guarantee in biopharmaceutical manufacturing is CHANGE. But custom designs and “discretionary upgrades” are often expensive, wasteful, inflexible, and not aligned with sustainability targets. Using risk assessments to understand current and future program requirements can help manufacturers avoid these pitfalls. By establishing a better baseline for process and facility design, risk assessment methodologies are the foundation of sustainable design.
A broader definition of risk
While identifying risks to patient safety is paramount, assessments like those highlighted in this article extend beyond that critical focal point, uncovering risks to operator health, supply chain reliability, delivery timelines, and more.
Cost-related risks are often targeted by risk assessments. Consider, for example, a manufacturer concerned about overspending on unnecessary maintenance and redundancy. A risk assessment identifies new elastomers, gaskets, diaphragms, and other small components as major drivers of maintenance costs. The manufacturer changes these components every year—always has, under the assumption of necessity. However, data gathered during a risk assessment may reveal opportunities to extend the life of these components through relatively low-cost or even money-saving improvements, thereby meaningfully reducing overall maintenance spending without putting product quality at risk.
3. How should manufacturers prioritize risks identified in a risk assessment?
Comprehensive risk assessment methodologies culminate in a risk index (RI), a calculation that is a function of a hazard’s severity and the likelihood the hazard will occur. A RI Matrix depicting 4-level likelihood and severity risk criteria is presented below.
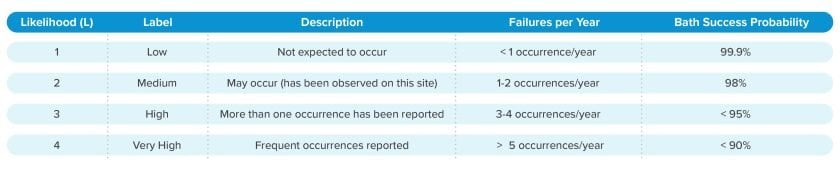
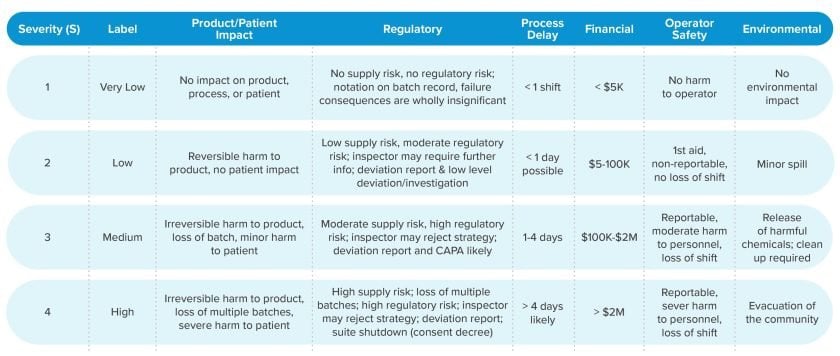
When risk represents harm to patient:
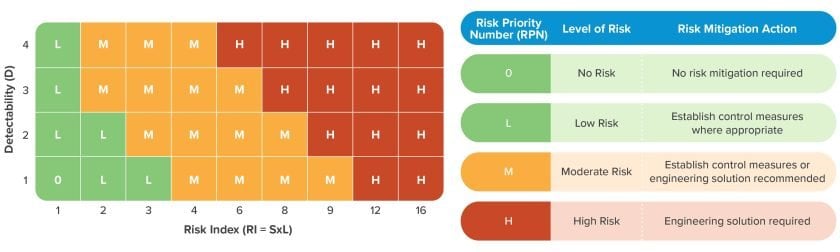
In biopharmaceutical manufacturing applications, patient safety is paramount. Risk to process and product is important, but regulatory agencies focus on harm to patients. Distinguishing between process safety and patient safety may therefore be a valuable practice. This is where detectability plays a key role.
Detectability: The ability to detect the existence or manifestation of a hazard.
Although the ability to detect a hazard or risk does not in itself alter the likelihood of occurrence, detection of a hazard prior to release of product to a patient (and subsequent harm to the patient) is helpful in making better decisions about risk acceptability.
Detectability can be quantified independently from other risk criteria. The timely detection of an out-of-specification product is an effective mitigation of risk to patient health.
Hazard
In the context of producing, controlling and distributing pharmaceuticals, a hazard is a real or potential condition, situation or agent that could cause harm to products, facilities or processes, and/or result in an adverse health effect in patients. Hazards may also damage systems (potentially resulting in the loss of a system) as well as equipment or the environment.
Harm
A harm is present when a hazard or risk could negatively impact a patient, operator, facility, or community due to a lack of ability to detect or monitor the risk element in a timely fashion.
The Risk Priority Number (RPN): is a function of Risk Index (Likelihood & Severity) and (Detectability). Together, these distinct measurements help design teams recognize and optimize two strategies for mitigating risk: reducing the probability of an issue in the first place and increasing the probability of detection should that issue persist. The RI and RPN should be assessed independently as some hazards that may be of acceptable financial risk could potentially represent unacceptable risk to patients. In CLARA, a separate matrix focused on patient safety may be useful.
Risk Priority Number (RPN): The result of the function of the risk index (RI = ƒ(likelihood, severity)) and detectability of the hazard or risk element. (RPN = ƒ( RI, detectability))

4. How are risk assessments evolving to address new manufacturing strategies and contamination challenges?
Innovation in the life science industry requires agility, flexibility, and visionary leadership. But for innovative ideas to drive transformative and patient-centric change in the way drugs are made, another ingredient is necessary: a culture of ongoing and rigorous risk assessment.
Without a systematic way to assess the impact of innovations on patient, operator, and business outcomes, manufacturing pioneers are unable to drive the industry forward. Risk assessment provides that mechanism, giving manufacturers the tools they need to responsibly adopt and adapt new processes, designs, and strategies for the commercial GMP environment.
Process closure is one of the most transformative of these new manufacturing strategies to emerge over the last decade, and risk assessment continues to play an integral role in its evolution.
Closed processing: a glossary
A system that isolates the process zone from its manufacturing environment and prevents the ingress of environmental contaminants during product contact.
A system that is subject to the contaminants in the manufacturing environment.
Open process where the process conditions mitigate the risk of contamination from the environment while the process solution or product solution is exposed to the environment prior to rendering it closed (for example, by sterile filtration). Examples of controlled conditions could include process duration, temperature, pH, bacteriostatic/bactericidal conditions, and viable/non-viable particulate loads.
The environment that includes all elements (such as raw materials, chemicals, products, contaminants, personnel, or waste) that may contact the process or product. In a closed system, the process zone is limited by the process zone boundary, which isolates the process zone from the manufacturing environment. In an open system, the process zone includes the manufacturing environment.
A set of controls that helps ensure the quality and safety of a product by identifying, monitoring, and mitigating potential contamination sources.
For more key terms related to closed processing, see the glossary included in BioPhorum’s Closure Playbook.
The benefits of process closure
Process closure answers a game-changing question: What if we eliminated the cleanroom?
Though they’ve been a standard component of the life science manufacturing facility for decades, cleanrooms are a flawed mechanism for controlling contamination, particularly given their heavy reliance on operational integrity. It takes just one mistake—a rushed gowning process, a door opened at the wrong moment, a fingernail puncturing a glove—to put entire batches at risk. This risk is exacerbated by the fact that detecting these deviations is almost impossible. In addition to operational integrity, manufacturers must also maintain energy-intensive utilities to meet the airflow, temperature, and humidity requirements of a cleanroom environment, driving up a facility’s operational costs and carbon emissions.
Process closure promotes a different contamination control philosophy. By transferring reliance from the cleanroom to the equipment, process closure frees manufacturers from the costs, complexities, and risks of controlling bioburden in an uncontrollable environment. That means the manufacturing process could, in theory, happen almost anywhere. Even regulators have acknowledged this possibility: in their 2016 revision of the Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients, the FDA states that “Where the equipment itself (e.g., closed or contained systems) provides adequate protection of the material, such equipment can be located outdoors.”
Who benefits when manufacturers untether themselves from the cleanroom by implementing closed processes?
Patients
The controlled and segregated environment of a closed processing system enables more effective contamination control, which in turn improves the consistency and quality of final drug products. And because production isn’t bottlenecked by time-consuming room changeovers, manufacturers may be able to accelerate delivery of the products that patients rely on.
Manufacturers
Closed processing allows for smaller, simpler facilities with open ballrooms, fewer airlocks, and less utility piping. This means faster project delivery for new facilities, which shrinks capital costs. Reduced maintenance needs and lower overall energy consumption may also reduce operational spending.
Closed processing also offers future flexibility. The heavily customized nature of a cleanroom-centric facility limits manufacturers’ adaptability, potentially leaving them with a very expensive paperweight as the industry moves on to new manufacturing models. Closed processing, on the other hand, opens the door to multi-product and multi-modal production or decentralized models that put production close to the patient’s bedside.
Operators
By segregating in-process substances within closed systems, manufacturers limit the risk of exposing operators to harm.
The environment
Manufacturers can significantly lower their greenhouse gas emissions and utility bills by eliminating or reducing the fans, boilers, chillers, and other utility systems that maintain the environment inside a cleanroom.
These benefits are driving a surge in the adoption rate of closed processing systems. For our annual Horizons: Life Sciences report, for example, our team surveyed manufacturers with mRNAs in their pipeline and learned that approximately 50% expect to implement closed processing within the next three years.
How will they get there? Applying a robust CLARA is part of that answer.
Closure Analysis Risk Assessment (CLARA)
Alongside its potential benefits, process closure presents several unique challenges. Closed systems must be designed, installed, and used correctly. The same applies to utility streams: the quality of Water for Injection (WFI), buffers, and other materials that feed a closed system must be commensurate with process requirements. There are vital procedural factors to consider, as well. For example, closed equipment may be opened for manual steps, such as assembling or connecting components, but must be closed again prior to product contact. Manufacturers must apply validated cleaning, sanitation, or sterilization steps to close those exposed systems.
There is no one-size-fits-all system or methodology to achieve process closure. Manufacturers must consider multiple possible approaches through the lens of their QRM strategy, using an appropriate risk management tool to monitor and document their approach. Designed to meet this need, CLARA will be formally presented in an upcoming chapter of BioPhorum’s Closure Playbook.
The CLARA methodology uses a hybrid HACCP and FTA to evaluate the isolation of process and product contact systems from contaminants in the surrounding environment. First, FTA visually identifies all sources of contamination in the manufacturing environment that could potentially reach the process zone and, ultimately, the process and product. Then HACCP systematically evaluates these potential sources of contamination and helps the risk assessment team identify solutions to mitigate these risks effectively.
Risk assessment teams can use CLARA to assess closed systems from installation through calibration, cleaning, sterilization, and operation, as well as all connected utility streams. If the closed system has vulnerabilities that might expose the process zone to the external environment, CLARA will find them. Using the pre-established risk criteria (likelihood, severity, detectability, risk index, and risk priority number), drug manufacturers can then prioritize each risk of breach of integrity of the process zone and closed process.
A successful CLARA will prove to the design team, project owners, and regulators that appropriate mitigations are in place to prevent the room environment from impacting the quality and reliability of the process and product within the closed system. The regulatory guidelines, in turn, have made it very clear that closed systems do not require classified cleanrooms, a key enabler of the future of fast, flexible, patient-safety-focused drug manufacturing facilities.
5. What goes into a successful risk assessment?
Risk assessment tools vary in complexity and require time and resources to use effectively, but they share a few general criteria for success.
A culture of collaboration and team engagement
Effective risk assessments rely on input from stakeholders across the organization, including end users who have direct experience with the systems and processes under scrutiny. People who bring different perspectives, levels of experience, and disciplinary knowledge should be involved, including input from the quality, manufacturing, validation, engineering, regulatory, and maintenance teams. Facilitators should endeavor to create an environment where all stakeholders feel free to speak candidly, identify issues they’ve noticed, and suggest mitigations that may help guide the risk assessment.
Access to good historical data
Reliable, up-to-date data will help risk assessment teams maximize tools like modeling and simulation. These tools generate the insights necessary to make proactive, targeted changes that support lower risk and better outcomes for patients, operators, and businesses.
A commitment to continuous improvement
Many risk assessment tools, like CLARA, are design tools—the earlier they’re applied in the design process, the more effective they become. But risk management doesn’t end once a facility is built or a process is established. It’s a continuous exercise that depends on regular reviews, feedback loops, and iterative adaptations as new opportunities or risks arise.
Risk assessments: accelerating responsible progress in biomanufacturing
In the context of the life sciences, the term “risk” typically evokes fundamental issues such as the safety of patients and facility operators, the reliability of production schedules, and the efficiency of resource allocation.
As successive waves of innovation shape this industry, another dimension of risk is rapidly gaining importance: the risk of failing to adapt as new technologies or manufacturing philosophies emerge or of rushing to implement a promising strategy without thoroughly and methodically evaluating its end-to-end implications.
Risk assessments, applied systematically and with scientific rigor, are a manufacturer’s most important defense against all types of risk. From shaping a strong contamination control strategy for a single production train to future-proofing a global manufacturing network, applying the right risk assessments at the right time can help manufacturers move forward safely, cost-effectively, and with confidence.
To strengthen and broaden your risk assessment toolkit, talk to our experienced assessment team today.