How the ATMP and cell and gene therapy industries can prepare to scale manufacturing operations safely, sustainably, and strategically.
Read More
Scale-up viral vector production
Get expert guidance to develop your viral vector manufacturing strategy.The steam locomotive. The Ford Model T. The lunar module. Each of these feats of engineering gave us access to places never before within reach.
Today, a different kind of vehicle is taking us to a place of immense possibility. The viral vector, a gene modifying platform, can act as a gene therapy and a key component of genetically modified cell therapies; and it’s opening the door to a world in which previously terminal diseases may have a cure.
But as viral vector-based therapies move beyond ultra-rare indications to target common conditions with larger patient populations, manufacturers need to overcome significant challenges. Just as Ford had to revolutionize the industrial factory to meet demand, today’s viral vector manufacturers must rethink GMP manufacturing in order to survive the journey from benchtop discovery to bedside medicine.
That means navigating a broad web of complex decisions and tradeoffs: which virus type to use, which cell line, which vector production technique, etc. There is no single “best” way to assemble these complex puzzle pieces, and even small changes can create big swings in terms of:
- The scalability of your facility and your process
- Your ability to close and automate production
- Your dependence on human operators
- Your material costs
- Your compliance strategy
In this article, we’ll examine these factors and explore the relationship between your lab-scale viral vector development pipeline and the cost, efficiency, and future flexibility of your commercial-scale facility. You’ll walk away with a plan for aligning today’s R&D decisions with tomorrow’s vision of a successful GMP operation, supported by a facility designed to accelerate your process, generate a sustainable ROI, and deliver more life-saving therapies to the patients who need them.
Viral vector manufacturing terms to know:
A viral vector is a virus that is deployed as a carrier to deliver genetic material into cells. In the process of making a viral vector, manufacturers disable its dangerous pathogenic features while taking advantage of its natural ability to penetrate cellular material.
A platform process is a standardized manufacturing approach that aligns equipment, protocols, and facility design in a harmonized system, enabling greater efficiency at scale. With a platform process in place, research teams have the guardrails they need to align their development efforts with the long-term vision for a commercial manufacturing operation.
The Pharmaceutical Inspection Co-operation Scheme (PIC/S) Annex 1 and the European Union’s revised Annex 1 (“Manufacture of Sterile Medicinal Products”), released in 2022, lays out the requirements necessary for manufacturers to validate their operation as aseptic. Compliance with Annex 1 criteria requires a robust contamination control strategy (CCS), which implicates a complex array of factors including equipment selection, personnel gowning and flows, material and product flows, and waste handling.
For manufacturers who are able to sterilize their bulk product via filtration, the Annex 1 “bubble” encompasses only the fill-and-finish operation at the culmination of the production lifecycle. For those who decide to forego sterile filtration due to significant yield loss, though, extending that bubble to include all downstream operations may be necessary, at a high cost.
AAVs offer versatility and improved tissue targeting compared to other options, making this a popular vector for in vivo genetic modification. Its payload is limited, though, and the DNA it delivers doesn’t integrate into a cell’s genome. As a result, it’s typically used in treatments that target non-dividing cells, like those in the nervous system.
These vectors share many characteristics with AAVs, with the distinction of carrying a genetic payload that’s several times larger.
LVs and RVs carry a large genetic payload, but unlike both AAVs and ADVs, their genetic material gets integrated into the host cell genome. This makes LVs and RVs a popular choice for companies pursuing ex vivo treatments involving dividing cells, such as CAR T cell therapy.
This non-mammalian vector type is a versatile, low-cost option—and because it’s only capable of replicating in insect cells, it reduces the biosafety burden that manufacturers face when designing their CCS.
This cell type requires a tissue or mesh surface as an anchor point in order to reproduce. As a result, it’s the most “spatially greedy” of potential cell lines, which has long-term implications in terms of facility footprint.
Cells of this type can reproduce in the growth medium without an anchor point. This allows manufacturers to leverage the scalable devices already well-established in other industries, like the stirred-tank bioreactors common to monoclonal antibody manufacturing.
This is the most widely used vector production technique. Think of a locked safe that will only open when two keys are turned, and each key is hidden in a different place. Some of the genes necessary for the formation of the viral vector are encoded in plasmids or other transgene elements, and some are in the cell line itself. The viral vector can only form when that specific host cell line is transfected with those plasmids, which happens once cells have grown to production volume.
Despite its popularity, this technique is problematic from a scale-up perspective, which we dive into later.
Rather than waiting for the cell line to reach production volume, this technique involves engineering cells during generation of the master stock. This means that as cells reproduce, so too does the vector.
While it can take significant development time to engineer a stable cell culture, the scale-up advantages are numerous and may be worth the upfront investment.
The challenge facing today’s viral vector gene therapy manufacturers
For patients, the prospect of viral-vector-based therapies expanding their reach to address more common diseases is promising. Already, that promise is becoming a reality: as of early 2023, eight gene therapies that rely on viral vectors have FDA approval and more than three times as many are in late-stage development. To prepare for this acceleration in development, the FDA recently expanded its cell and gene therapy regulatory unit, which it expects will approve between 10 and 20 applications annually by 2025.
Manufacturers with viral vectors in their development timeline are asking what challenges must they overcome to reach that point of regulatory approval and commercial success in the next few years.
To understand the answer to this question, let’s examine one of the main reasons that viral vector manufacturers may find themselves facing bottlenecked processes, increased cost of goods (COGs), and a sluggish return on capital investments as they scale towards commercial production: they aren’t starting with a well-established, standardized platform.
Without standardization, scalability is off the table
With the industry still so new, viral vector manufacturers are looking to other markets for well-established tools and techniques. To some degree, this has proven useful; manufacturers with a suspension-capable viral vector platform can leverage the scalable processes behind single-use monoclonal antibody manufacturing, for example. But viral vector manufacturers with an adherent platform may find themselves locked in a process that recalls the early days of biotech when processes hampered scalability.
In both scenarios, it can be difficult for viral vector innovators to know when, how, and if to invest in capital infrastructure. Where should they put their attention to ensure the future scalability and commercial readiness of their manufacturing process?
The answer: Plan for commercial scale from day one
To address this challenge, plant the seeds of a robust, scalable platform process early on by building alignment between your R&D priorities and the business case driving your commercial objectives. That means recognizing that each decision made during small-scale process development needs to work in a regulated, commercial environment as well—it’s not enough to simply get the job done at the lab bench. Approaching process development this way may require more time, but that upfront investment will pay for itself as you scale to the large volumes required to meet patient demand.
Let’s examine what this “commercial state of mind” looks like at each stage of the manufacturing process, and how a platform-based approach to early process development will position you to operate a smaller, simpler, more efficient commercial facility in the future.

How does upstream processing impact facility design?
Rather than hitting the “easy” button and sprinting ahead with a cell line that will quickly get you to your immediate next step, it’s important to consider the end-to-end implications of your upstream viral vector manufacturing approach.

Vector type: AAVs, ADVs, LVs, RVs, and BEVs
It’s important to understand how your chosen vector’s physical characteristics, such as its size and stability, will impact the scalability of your process and your approach to facility design and optimization.

Your vector selection has a cascading effect on other key decisions, which in turn impacts your facility use case. It’s important to weigh the benefits and tradeoffs of your vector type before you’re committed to it, and ensure that it aligns with your commercial scale-up goals. Adeno-associated viruses (AAVs) are the current vector of choice for the majority of developers, but retrovirus and lentivirus applications are also popular (for more on each virus type, see our “Terms to Know” section). Choosing the right fit for your project takes careful consideration.
For example, baculovirus expression vectors (BEVs) are capable of growing in a suspension culture without complex engineering involved, which opens the door to a facility that’s smaller and more efficient than one designed for adherent cultures. But for all of their attractive qualities, BEVs introduce unique purification challenges which could impact the facility’s downstream layout.
Your vector’s suitability for sterile filtration will impact the complexity of your facility. If your virus type can withstand sterile filtration without significant yield loss, then you’ll be able to limit the expense and regulatory burden of an aseptic environment.
If, however, your virus type is too large to pass through a sterile filter, you may need to expand your aseptic environment further upstream to ensure sterility—which brings with it the expense of higher background classifications, more airlocks and gowning procedures, and advanced equipment designed to support aseptic manufacturing. We’ll talk more about this scenario in our examination of downstream manufacturing, below.
Your choice of cell growth technique
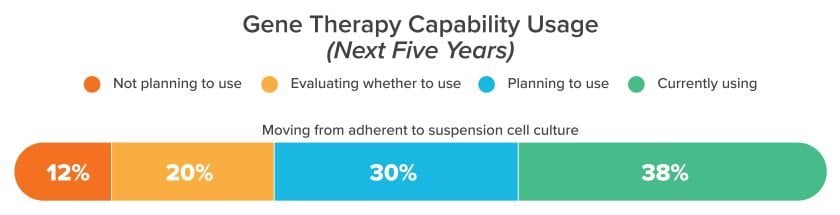
Adherent cell lines can accelerate early clinical development phases, but their poor scalability is a disadvantage as manufacturers transition to commercial manufacturing. That’s driving a growing number of R&D teams towards suspension-based approaches, as we saw in our 2022 industry survey of nearly 500 respondents (Horizons: Life Sciences). The bottom line: it may take longer to develop suspension-capable cell cultures, but this early investment will open the door to scalable and validated GMP technologies.
Adherent cell line (also called “anchorage-dependent”)
You’ll need a lot more room. Adherent cell lines need surface area to grow. While it’s possible to adapt stirred-tank bioreactors for this use, this requires complex engineering—especially at large commercial volumes.
Instead, most adherent cells are cultured in multi-plate and fixed-bed bioreactors, leaving you with islands of activity that are much more difficult to scale up than to scale out. The result is a larger, less densely utilized viral vector manufacturing facility.
You’ll need additional warehousing and staging areas. It takes a significant volume of pre-sterilized plasticware and purchased media to support adherent cell culture growth. You’ll need adequate warehousing to store that material, as well as a facility plan for staging, transporting, and disposing of it.
As your substrate containers grow, so does the strain on your human operators. Adherent cell lines require heavy use of personnel to undertake cell washing and expansion operations. Not only will these manual steps drive up your operational costs, they may also expose you to a greater risk of human error.
This production approach resists automation and process closure. Operators will need to manually facilitate and perform cell washing and expansion operations. This requires a larger skilled workforce, which in turn necessitates larger ancillary spaces such as locker rooms, gowning areas, and so on.
Suspension-capable cell line
Manufacturers can scale up. Suspension-capable cell lines reproduce in the growth medium without the need for anchor points. This frees manufacturers from having to accommodate adequate surface area. Using commercially available single-use bioreactors, manufacturers can plan a linear scale-up approach from 25 to 500 L—some viral vector manufacturers with suspension-capable cell lines are even planning for 2,000 L units.
This scale-up pathway may not be entirely free of challenges. Some suspension-capable cell types tend to clump at high densities, for example. Solving these challenges will require further process development.
Suspension-capable cell cultures open the door to well-understood technologies. To produce viral vectors using a suspension-capable culture, manufacturers follow many of the same steps that producers of monoclonal antibodies or therapeutic proteins have had in place for decades.
Instead of relying on complex engineering or individually optimized processes, this means you can leverage existing knowledge to establish a scalable platform approach that maximizes the benefits of closed and automated processing technologies. This is the key to a more efficient facility.
A transition from adherent to suspension-capable in the future requires forethought and planning. Many manufacturers plan to meet their speed-to-market milestones by launching with an adherent cell line culture while developing a suspension-capable alternative in parallel for a future launch.
This degree of complex engineering may take significant time. It’s also risky; by committing to an adherent cell line before knowing for certain if it can be adapted for suspension (or if its suspension-capable version will be productive enough to justify the time and cost invested to develop it), you’re putting your capital investment on the line. Spending upfront resources to understand the journey to suspension-capable cell lines before facility build-out could pay off in the end.
Your choice of vector production technique
Broadly speaking, there are two main platforms that the majority of viral vector manufacturers use, each with its own limitations and advantages: helper/packaging cells and stable/producer cells.
Their differences come down to how they propagate a specific vector, which has far-reaching implications in terms of the space and resources required within the manufacturing facility.
Helper cells are popular for lab-scale viral vector development, but as manufacturers look toward commercial-scale up, more of them are moving toward stable producer cell lines. That may come down to the fact that helper cell lines rely on large volumes of raw materials, which can drive up costs and expose manufacturers to supply-related delays.

Using helper cells as your vector production technique
You can run multiple production streams in parallel. This is a plus. Because helper-based cell lines aren’t transfected until they reach their target volume, you can run parallel trains of cell culture within the same cell expansion suite—and you can leverage the same equipment by staggering growth cycles. This invites a highly dense use of facility footprint. With good planning, this pro may counterbalance any potential cons.
You will need to invest in large volumes of raw material. In helper cell lines, the transgene conversion happens only once cells have grown to production volume. At commercial scale, that means acquiring and handling enough plasmid (or similar element) to transfect potentially 500 L of cell culture or more.
This can raise your upstream raw material COGs. If you rely on a CDMO to supply that raw material, you may also need to plan for long lead times. It’s not uncommon to wait a year or more for delivery of a custom transgene plasmid.
You may be limited to scaling out. Manufacturers with helper cell lines in development typically rely on transient transfection to deliver the genetic payload. Compared to stable producer cells, this technology promises a bigger bang for your buck in terms of protein expression.
On the other hand, transient transfection works best in smaller bioreactors, which makes it a difficult technology to scale up. For example, two 200 L single-use bioreactors may offer better dynamics (and therefore a higher throughput) than one 500 L bioreactor.
Your purification process may be more complex. Unlike stable producer cells, helper cells have a limited lifecycle. Once they produce a vector, they are lysed—at which point they are considered a contaminant that must be removed via filtration. That adds a layer of complexity to your downstream process, necessitating further process segregation and more personnel.
Using stable/producer cells as your vector production technique
You’ll invest far less in raw material. Rather than waiting for the cell line to grow to production volume (as in the case with helper cells), these cells are transfected from day one. This transfection is stable, not transient—as cells reproduce, their modified genome reproduces with them, transfecting future generations.
This is good news in terms of upfront costs, because manufacturers only need enough plasmid to transfect their initial cell stock. As your cell line grows to production volume, your transgene element essentially grows with it. There are questions about the long-term effects of stable transfection on cells, though.
Your purification process may be more streamlined. Helper cells must die and break apart to release the viral vector—a process that necessitates complex cell lysing and clarification steps to remove DNA contaminants from the cell broth. In contrast, stable cell lines express the vector extracellularly. This eliminates the need for depth filtration of cell debris, enzymatic digestion of residual DNA, and ultracentrifugation to remove empty capsids. The result is a simpler facility in terms of downstream operations.
You may need to invest in a longer upfront development timeline. The road to a stable producer cell line will not necessarily be a smooth one. For example, your modified cell line may result in cell death due to cytotoxic byproducts or some other factor. Plan for enough upfront resources to address these challenges and unlock the benefits described above.
You may need to segregate the entirety of your production streams. Because they are engineered on day one, stable cell lines introduce the risk of viral cross-contamination from initial vial thaw.
To manage that risk, manufacturers may need to design facilities for total segregation, including segregated cell expansion processes in GMP clean rooms—which brings with it the added cost of expanded mechanical systems, gowning, cleaning and monitoring, validation, and so on.
Raw material requirements: insourcing or outsourcing?
The upstream decisions discussed in this section all impact the volume, complexity, and cost of raw materials, which in turn determines the way you use your facility.
- Raw materials may include plasmid DNA, media, virus seed stock, animal-derived serum (for growing adherent cell lines), and so on.
- Sourcing these GMP materials is a big driver of upstream COGs, and a cause of delays (industry shortages, long wait times)
- As you scale from benchtop research to commercial-scale production, the volume of available raw materials must scale with you. Without advanced planning, this issue can cause a serious production bottleneck.
In the midst of these complexities, manufacturers face an important question: in the long run, will they be better off investing in the capacity to produce raw materials in-house? Or does it make more sense to rely on third-party suppliers? Or is a hybrid of both options the best way forward?
The answer is as complex as the question—and it takes a clear end-to-end perspective of your whole facility master plan and the dynamics of the marketplace around you to arrive at a tailored answer.


How does downstream viral vector processing impact facility design?
At the beginning of the downstream process, viral vectors are components in a crude broth. By the time that process finishes, they’re ready for final formulation and delivery to patients. What happens between these two points is a ballet of open and closed processing. Many of the choices made to support upstream viral vector processing have a cascading effect on this downstream choreography, such as the vector’s physical characteristics (which can complicate filtration steps) or the use of serum to supplement cell culture media (which increases the purification burden).
- Comply with stringent regulatory standards. If aseptic processing is required, meeting the appropriate regulations without comprising production efficiency is especially challenging.
- Align throughput capabilities with demand, which means developing and implementing a flexible high-volume commercial scale-up strategy.
To meet these criteria, manufacturers need a flexible facility that’s designed and optimized for their unique downstream processes. Below, we’ll explore a few of the challenges and solutions we’ve seen in the field, and what they mean at a facility level.
Open vs. closed processing: a fine balance
The upstream viral vector manufacturing process is necessarily closed to protect the cell culture’s environment. As that process moves further downstream, though, maintaining process closure becomes more challenging. Successively smaller batch sizes could force you away from closed, automated equipment designed to process large volumes of material and towards benchtop solutions typically reserved for open, lab-scale operations.
Vendors are aware of the need for robust, commercial-ready downstream equipment that supports closed and automated processing for small batch sizes, and innovative solutions are emerging all the time. For now, though, many manufacturers face the prospect of making smaller-scale open technologies work in a controlled GMP environment.
Harvest, clarification, and concentration
You’ll need to plan for open processes and corresponding room segregation. Clarification steps, which are usually performed in the same suite where vectors are propagated, are generally open; that means they must be segregated from downstream purification steps to prevent cross-contamination.
Upstream decisions impact the downstream bioburden control strategy. If you relied on helper cells to propagate vectors during upstream processing, you may need a facility that’s sized and optimized to support cell lysis and viral filtration. You’ll also need to anticipate the purification burden generated by these steps; cell lysis, for example, uses a detergent solution to break open cells, releasing the vector while also polluting the cell broth with impurities, contaminants, and other cellular debris.
Purification and concentration
Smaller yields mean smaller equipment. Purification typically involves affinity chromatography as a first step, followed by ion exchange chromatography to separate full from empty capsids, and finally ultrafiltration to improve concentration.
By the time this process reaches its last chromatography step (usually after two or three columns), your viral yield may have fallen so significantly that commercially scaled GMP chromatography skids are no longer suitable. That leaves you with small, benchtop skids which may not be suitable for a GMP environment. Their construction materials may be incompatible with GMP cleaning routines, for example, or they may not support the required level of automation and process control.
You may face the challenge of adapting chromatography steps to meet aseptic processing criteria. If you’re implementing a fully aseptic downstream process, you’ll need to sterilize your chromatography columns—an all-new challenge with no well-established solutions.
If you pursue ultracentrifugation, you need to plan for it in your facility layout. It may take significant development time to engineer an ion exchange chromatography process that’s optimized for your particular serotype. To accelerate their speed to market, some manufacturers will take a different route, choosing instead to separate full from empty capsids using large-volume ultracentrifuges.
What they save in time may cost them in terms of space and scalability, though. Ultracentrifugation relies on open and manual processes, which are more suitable for lab-scale processing than for processing inside a GMP facility. If you take this path, you’ll need to plan additional space for all of the infrastructure that comes with higher background classification and appropriate gowning, airlocking, and so on—as well as the need for manual operators.
Sterilization
If sterile filtration isn’t possible, you may need to rethink your downstream process from an aseptic perspective. Ideally, manufacturers rely on sterile filtration to remove potential microbes from their bulk product. In this scenario, only the final fill-and-finish environment is subject to the rigors of aseptic processing (as defined in the recently revised Annex 1 documentation).
If your virus vector is large and enveloped, though, it may sustain significant yield losses during this sterile filtration step. That will drive your COGs exorbitantly higher, hurting your product’s commercial prospects. In this case, you may find yourself in a difficult situation, forced to expand that Annex 1 boundary to encompass all harvesting and purification steps.
That introduces a host of complications. You’ll need a process and a facility that’s designed and validated for downstream aseptic processing, which involves complex resource planning, mechanical infrastructure, and material handling protocols, each at a high cost. You’ll need an isolator or a biosafety cabinet to perform open steps. Single-use sterile process components will be necessary. And you’ll need a solution for your chromatography systems, which aren’t historically designed to support aseptic processing.
These burdens are significant enough to justify sterile filtration in many scenarios, despite its impact on vector yield. This decision could have implications for the end-to-end manufacturing process; for example, it puts pressure on upstream processes to improve the cell culture titer, thereby helping to offset downstream yield loss.

Fill-and-finish: right-sizing for viral vectors
For manufacturers delivering large volumes of a therapy to market (such as monoclonal antibodies), it may make sense to outsource fill-and-finish operations to a partner with automated filling lines and other specialized capabilities.

For viral vector manufacturers, though, developing in-house fill-and-finish capabilities could be a better investment than outsourcing.
That’s because final product volumes are typically quite low for viral vector manufacturers, which means the large-volume capacity of a CDMO may be overkill. Manufacturers could instead meet their own needs by hand-filling vials inside a biosafety cabinet or by using a small isolator with automated filling capabilities.
This in-house approach can give manufacturers better control over product stability throughout the filling process, and it avoids the logistical and regulatory headaches involved in transporting bulk products to another facility.
A manual filling process may not be adequate for some viral vector manufacturers. To avoid bottlenecks, those with larger batch volumes or plans to expand may consider adding an automated filler to their internal infrastructure. This means operating a Grade A isolator inside a Grade C background, which will require more design consideration, facility infrastructure, and upfront capital cost.
Some vendors now offer pre-designed modular filling isolators to minimize the design input required to expand internal fill-and-finish capacity, though these options introduce a few important disadvantages such as poor future adaptability and a limited section of packaging options.
With in-house fill-and-finish capabilities comes elevated testing requirements.
Another factor to consider when evaluating the viability of an in-house fill-and-finish operation is the quality control burden. In addition to regular in-process testing, manufacturers with their own fill-and-finish process will need to conduct extensive release testing, which in turn means larger, more complex QC labs. The upfront capital cost required to plan adequate lab space may be justified by the long-term rewards of controlling your entire process train internally.
What’s next for viral vector manufacturing?
The eight viral-vector-based gene therapies currently in the market are the crest of a wave. With nearly 150 more in late-stage development or Phase II trials, that wave is growing rapidly—and with it, new solutions to the challenges of standardizing and scaling viral vector processes are emerging every day. Already, advances in artificial intelligence and smart data analytics are opening the door to accelerated development pathways, improved vector yield, and more efficient and automated manufacturing approaches at scale.
To stay agile and ready for these new innovations and opportunities, viral vector manufacturers need facilities that are flexible, standardized, and designed to generate value long into the future. A partner can help; while you’re focused on developing a breakthrough vector, we can marshal our experience of designing and building hundreds of advanced therapy projects to help you plan your own unique way forward.
When you’re ready to discuss your viral vector manufacturing scale-up strategy, let’s talk.